由2.5-二甲氧基氯苯合成4-氯-2.5-二甲氧基苯胺研究
2,6-二溴-4-三氟甲氧基苯胺的合成

制工艺去除 , 得到高纯度 的 26 二溴 一 三氟甲 ,一 4一 氧基 苯胺 。因此 , 本工艺 合理 , 操作 简单 , 成本 低 , 非
常适 合 工业化 生产 。
使用有机溶剂二氯 甲烷做溶剂进行溴化时 , 反应从 2 实验方法简介 溴取 代 逐渐进 行 多溴取代 , 同时 , 生成 的溴 化氢 直 2 1 产 品分 子式 . 接成盐并大量析 出, 形成悬浮液, 使搅拌非常困难 ,
B
Br
OC 3 F
2 2 主 要反应 方程 式 .
但是 , 乙酸用量 很大 , 价格 昂贵 , 不适合 工业 化生 产【 。如果使用水做溶剂进行溴化时 , 3 】 虽然反应也
能进行 , 但是 反应 物 成 团状 , 反应 进 行 的很 不 均 匀 ,
昔 B B r
OCF
氢还 原 为溴 素 , 直接循 环利用 , 低 了生产成本 。同 降
反应 结束 后 , 残 留 的溴 素 和生 成 的溴化 氢 必 须 进 对
时, 在反 应过 程 中 , 乎 不 形 成一 溴 取 代 的副产 物 , 几 可得 到 高收 率 的 26一二溴 一 , 4一三 氟 甲氧 基苯 胺 , 产物 中虽含有 微 量高 沸 点 有 色物 质 , 可 以通过 精 但
中间体 , 它 可 生 产农 药 杀 菌 剂 … ( 噻 氟 菌 胺 ) 用 如 和农 药 除草剂 等 。 通常, 苯胺 类 的溴 化 是在 有 机 溶 剂或 水 中进 行 反应 , 是 , 但 由于使 用溴素 , 反应 过程 中生成 溴化氢 ,
组 成 的两相 溶剂 中 , 三氟 甲氧基 苯胺 作为 原料 , 对 与
溴化 氢 容易 回收 , 以循 、 二氯乙烷、 一氯苯、 二氯苯等。 在实验过程 中, 使用的水 和有机溶剂 的用量 比 在 5~ 5 9 5 水/ 9/ 5— ( 有机溶剂 ) 之间较好 , 最好 在 1 — 09 1 之间。实验 中使用的溴化剂可 以单 0 9/0— 0
2-氟-4,5-二氯硝基苯的合成

2-氟-4,5-二氯硝基苯的合成蒋锐;董燕敏;杨金会;赵天生;陈兴权【摘要】以4-氟苯胺为原料,经乙酰化、氯代、水解、重氮化-桑德迈耳以及硝化合成了2-氟-4,5-二氯硝基苯.乙酸酐为乙酰化试剂合成4-氟乙酰苯胺,收率为95.9%;4-氟乙酰苯胺再经SO2Cl2氯代反应,合成2-氯-4-氟乙酰苯胺,收率为79.1%;2-氯-4-氟乙酰苯胺水解合成2-氯-4-氟苯胺,收率为73.6%;2-氯-4-氟苯胺经重氮化氯代制得3,4-二氯氟苯,收率为48.0%;3,4-二氯氟苯经硝化制得目的产物2-氟-4,5-二氯硝基苯,收率74.9%.过程的总收率为20.1%.%4,5-Dichloro-2-fluoronitrobenzene was prepared from 4-fluoroaniline through a route, namely acetylation, chlorination, hydrolysis, diazotization-Sandmeyer, and then nitrification. Firstly, 4-fluoroaniline reacted with acetic anhydride. The yield of the obtained 4-fluoroacetanilide reached 95. 9%. 4-Fluoroacetanilide converted to 2-chloro-4-fluoroacetaniline through sulfuryl chloride. The yield of the obtained 2-chloro-4-fluoroacetaniline was 79. 1%. 2-Chloro-4-fluoroaniline could be prepared from 2-chloro-4-fluoroacetaniline with the yield of 73. 6%. 3,4-Dichlorofluorobenzene was prepared by diazotization-Sandmeyer with the yield of 48. 0%. Finally, 4,5-dichloro-2-fluoronitrobenzene was prepared by nitrification with the yield of 74. 9%. The total yield of 4, 5-dichloro-2-fluoronitro-benzene by this route was 20.1%.【期刊名称】《精细石油化工》【年(卷),期】2013(030)001【总页数】5页(P12-16)【关键词】2-氟-4,5-二氯硝基苯;中间体;合成【作者】蒋锐;董燕敏;杨金会;赵天生;陈兴权【作者单位】常州大学石油化工学院,江苏常州213164;常州大学石油化工学院,江苏常州213164;宁夏大学能源化工重点实验室,宁夏银川750021;宁夏大学能源化工重点实验室,宁夏银川750021;常州大学石油化工学院,江苏常州213164【正文语种】中文【中图分类】TQ246.3+82-氟-4,5-二氯硝基苯是一种重要的医药、农药中间体。
硝化

硝化一、填空题1、衡量混酸硝化能力的两个重要参数是和。
2、工业上使用的主要硝化剂有等。
4、制备多硝基化合物或硝化产物难以进一步硝化的间歇过程,采用方式加料。
5、分离出废酸的硝化产物(或酸性硝基苯)中,还含有少量无机酸和酚类等氧化副产物,可通过方法使其变成易溶于水的盐类等而除去。
7、解离萃取法是以作萃取剂处理中性粗硝基物。
10、稀硝酸通常用于某些容易硝化的芳香族化合物,如的硝化,硝基主要进入羟基、烷氧基或酰氨基的对位,对位被占时则进入邻位。
11、当苯环上同时存在羟基(或烷氧基)和醛基时,采用的方法,可保护醛基不受影响。
12、亚硝化剂在亚硝化的反应中的活性质点是。
13、为安全起见,硝化反应器上应安装。
二、选择题3、混酸的脱水值越大,则混酸硝化能力()A 越强B 中等C 越小D 无法确定4、稀硝酸硝化活性质点是()A NO2+B H2NO3+C NO+D H2NO2+5、苯、氯苯或甲苯等的混酸硝化就是典型的非均相硝化反应。
研究表明, 非均相混酸硝化反应,主要是在()进行A 酸相和两相界面B 酸相C 有机相D 两相界面6、烷基苯与亚硝基硫酸及硫酸形成有色络合物(如C6H5R·2ONOSO3H·3H2SO4),使硝化液颜色往往会发黑变暗。
()最容易生成此络合物。
A带有长侧链的烷基苯 B短侧链的烷基苯 C 不带任何取代基的苯 D带有吸电子基的烷基苯7、工业上往往采用多个锅式反应器串联(简称多锅串联)的方法实现连续硝化。
连续硝化过程适用的加料方式是()。
A 正加法 B并加法 C反加法三、判断题1、在有机溶剂中的硝化剂的活性质点也不是NO2+,而是NO2+-OH2或NO2+-OAcH+等质子化分子,可看成是在H2O或HOAc分子上载有NO2+活性质点,其反应活性与形成NO2+的难易程度有关。
2、苯、氯苯或甲苯等的混酸硝化是典型的非均相硝化反应,主要是在酸相进行。
4、当苯环上带有供电子基时,硝化反应速度快,在硝化产物中常常以邻、对位异构体为主。
精细单元反应复习

• 3. 氯化深度也称为 ,它是 指 。 • • 4. 以硫酸为磺化剂的反应是一个 反应, 即 在较稀的硫酸存在下发生水解 ,且 浓度越高,反应速度越快。
• 1. 什么是碱性?什么是亲核性?二者之间 的关系如何? • • 2. 请说明三氧化硫磺化的特点。 • 3. 取代基的电子效应包括哪些内容?
• 1. (1)碱性指得质子的能力。 • (2)亲核性指对碳正离子的亲和性的大小。 • (3)亲核性与可极化度有关,当可极化度影响不明显, 碱性起主要作用时,碱性越强,亲核性越强。 • • 2. 优点 • (1)不生成水、无废酸; • (2)磺化能力强、反应速度快; • (3)磺化剂用量省,接近理论量; • (4)避免分离产品,产品质量高,杂质少; • (5)生产效率高。 • 缺点 • 反应剧烈,不易控制。 • 3. 诱导效应、 共轭效应、 超共轭效应
• 7、芳环上亲电取代卤化时,反应历程属于亲电取代反应, 活泼的进攻质点是氯正离子,以硫酸为催化剂,进攻质点 产生的过程是: 、 • 。 •
• 1 简述由对硝基甲苯制备以下芳磺酸的合 成路线、磺化的主要反应条件。 • (1) 2-甲基- 5-氨基苯磺酸。 • (2) 5-甲基- 2-氨基苯磺酸。 • (3) 5-甲基- 2-氨基-4-氯苯磺酸。 • 2 写出对硝基氯苯制取5-氨基-2-甲氧基苯磺 酸的方法。
• 8. 写出丙烯的氯化水解制1,2,3-丙三醇的工艺 过程。 • • • • 9. 乙酐分别与苯胺或甲苯相作用,可发生哪些 类型的反应,可得到哪些制品? • •
• 10. 简述由萘制备4-氨基-1-萘磺酸的合成路 线和工艺过程 • 11. 写出制备2-氨基-4-硝基苯甲醚的合成路 线和主要工艺过程。 • 12. 写出乙烯制备氨基乙酸的合成路线和各 步反应的名称。
AlCl_3催化对位取代苯胺与丙烯腈加成反应的研究

2009年第29卷有机化学V ol. 29, 2009第4期, 643~647 Chinese Journal of Organic Chemistry No. 4, 643~647* E-mail: zhaoying42@Received March 23, 2008; revised August 20, 2008; accepted November 25, 2008.湖南省科技厅科研基金(No. 2007FJ4156)资助项目.644有 机 化 学 V ol. 29, 2009在芳香胺的邻位、对位和间位取代苯中, 对位取代苯胺易与丙烯腈发生加成反应生成相应的N -氰乙基对位取代苯胺(简称单氰乙基物, 以下同). 但是, 生成的单氰乙基物要继续进行氰乙基化生成N ,N -二氰乙基对位取代苯胺(简称双氰乙基物, 以下同)则因空间阻碍而较难, 需要催化效率更强的催化剂, 但有关这方面的报道很少, 仅见Braunholtz 等[4,11]以HOAc-CuCl 催化体系合成N ,N -二氰乙基对氯苯胺, 收率为20%, Gimbert 等[12]以Bu 3P 催化合成N ,N -二氰乙基对硝基苯胺, 收率为71%, 本课题组[13~16]曾在合成N ,N -二氰乙基苯胺研究中发现AlCl 3对该类加成反应具有非常高的催化活性. 本研究继续采用无水AlCl 3催化一系列对位取代苯胺与丙烯腈发生加成反应合成相应的单氰乙基物和双氰乙基物, 并对合成的5种新的双氰乙基物进行物性和结构表征. 对位取代苯胺与丙烯腈加成反应方程式如Eq. 1.1 结果与讨论1.1 单氰乙基化反应相对于邻位取代苯胺、间位取代苯胺的单氰乙基化反应(单氰乙基化反应是指氨基上的一个氢与丙烯腈加成生成相应的单氰乙基物), 对位取代苯胺是一个没有空间阻碍的加成反应. 在无水AlCl 3催化下, 当AlCl 3加入较多, 丙烯腈过量较多, 反应温度较高, 反应时间较长的条件下, 易产生双氰乙基产物, 因此, 控制好这些反应条件尤为重要. 表1是我们优化的对位取代苯胺进行单氰乙基化反应的实验结果. 序号1~4为具有供电子基团的烷基、烷氧基等取代基, 单氰乙基化反应较易进行, 所以无水AlCl 3的使用量较少(2%, 质量比, 以下同), 反应温度较低(50~60 ℃), 其中甲氧基、乙氧基等烷氧基比甲基、乙基等烷基供电性更强, 反应时间要短, 反应温度要低. 氯原子诱导效应大于其供电子共轭效应, 使苯环上电子云密度降低, 所以, 对氯苯胺的反应活性比对甲基苯胺、对乙基苯胺要弱, 无水AlCl 3的使用量相对要多(10%), 反应时间相对要长(4 h), 反应温度相对要高(65~70 ℃). 序号6~7为具有吸电子基团的硝基、磺酸基, 较带烷基、烷氧基等供电子基团芳香胺的单氰乙基化反应要难得多, 即使无水AlCl 3的使用量更多, 反应时间更长, 反应温度更高, 也难以获得高收率单氰乙基产物. 其中N -氰乙基对硝基苯胺的收率仅21% (NMR 计算收率), 对氨基苯磺酸没有发生氰乙基化反应(分离产物的IR 表明无CN 基的吸收峰). 为降低双氰乙基产物的产生, 提高单氰乙基反应的选择性, 我们尝试采用加入一定量NaOAc 作助催化剂来降低AlCl 3的催化活性. 序号1~4与序号8~11的对比实验表明, 在反应时间延长后, 对甲基苯胺、对甲氧基苯胺、对乙基苯胺、对乙氧基苯胺和相应的双氰乙基产物的含量均降低, 单氰乙基产物的含量和收率均明显升高. NaOAc 之所以具有提高单氰乙基反应选择性的效果, 这可能是醋酸根上的氧原子与AlCl 3上Al 原子结合抑制了AlCl 3瞬时间强的催化活性, 避免了因剧烈催化作用而产生N ,N-二氰乙基对取代苯胺. 这与AlCl 3和许多有机碱性物组成的离子液来调节酸性的原理可能是一样的[17].表1 对位取代苯胺的单氰乙基化反应Table 1 Monocyanoethylation reaction of p -substituted anilines a产物含量b /% 序号 芳香胺 催化剂 催化剂/% 时间/h 温度/℃单氰双氰收率c /%1 p -CH 3C 6H 4NH 2 AlCl 3 2.0 4 55~60 92.5 2.5 89 2 p -C 2H 5C 6H 4NH 2 AlCl 3 2.0 4 55~60 91.7 3.0 88 3 p -CH 3OC 6H 4NH 2 AlCl 3 2.0 3 50~55 92.1 3.5 89 4 p -C 2H 5OC 6H 4NH 2 AlCl 3 2.0 3 50~55 92.0 2.8 88 5 p -ClC 6H 4NH 2 AlCl 3 10.04 65~70 93.5 0.7 906 p -NO 2C 6H 4NH 2 AlCl 3 20.0 12 80~85 — — 21d7 p -SO 3HC 6H 4NH 2 AlCl 3 20.0 12 80~85—— 08 p -CH 3C 6H 4NH 2 AlCl 3/NaOAc 4.0/2.0e 8 55~60 97.6 0.3 94 9 p -C 2H 5C 6H 4NH 2 AlCl 3/NaOAc 4.0/2.0 8 55~60 96.7 0.5 93 10 p -CH 3OC 6H 4NH 2 AlCl 3/NaOAc 4.0/2.0 8 55~60 97.5 0.4 94 11p -C 2H 5OC 6H 4NH 2 AlCl 3/NaOAc 4.0/2.0 855~60 97.1 0.4 93a对位取代苯胺用量为0.60 mol, 丙烯腈为0.66 mol, 芳胺为固体的加乙腈20 mL 作溶剂; b 系反应终点用高压液相色谱检测的含量; c 分离收率; d NMR 计算收率; e 4.0/2.0是指AlCl 3/NaOAc 的质量比.No. 4 赵莹等:AlCl3催化对位取代苯胺与丙烯腈加成反应的研究645可见, 无水AlCl3是催化对位取代苯胺与丙烯腈单氰乙基加成反应的一种高效催化剂, 控制丙烯腈和AlCl3的用量、反应时间和反应温度, 可以获得高选择性、高收率的单氰乙基加成产物; 而加入一定量的NaOAc作助催化剂, 则有利于提高单氰乙基反应的选择性和单氰乙基产物的收率.1.2 双氰乙基化反应对位取代苯胺单氰乙基化反应后, 再与丙烯腈进行氰乙基化形成双氰乙基物时存在空间阻碍, 使加成反应难于进行, 有关芳香胺的双氰乙基加成反应(双氰乙基化反应是指氨基上的两个氢与丙烯腈加成生成相应的双氰乙基物)报道很少. 我们采用加入较多的无水AlCl3、较多的丙烯腈、提高反应温度和延长反应时间较好地解决了这个问题. 表2是优化的对位取代苯胺进行双氰乙基化反应的实验结果. 与单氰乙基化反应一样, 烷基、烷氧基等具有强供电子基团的芳胺双氰乙基化反应较易进行; 而氯原子的诱导效应大于供电子共轭效应, 使苯环上电子云密度降低, 无水AlCl3的使用量相对要多, 反应时间相对要长, 反应温度相对要高. 序号1~4与序号6~9的实验结果对比表明, 由AlCl3-ZnCl2构成的催化体系比只用AlCl3催化具有更好的催化效率. 这是因为在AlCl3中加入另一Lewis酸可以构成超强酸催化剂, 如AlCl3-CuCl2, AlCl3-CuSO4催化体系就是固体超强酸催化剂[18]. 我们采用了AlCl3-Cu(AcO)2•3H2O, AlCl3-CuCl2, AlCl3-CuSO4, AlCl3-FeCl3, AlCl3-ZnCl2等五种二元催化体系研究该类反应, 发现AlCl3-ZnCl2催化体系具有更高的催化效率. 其原因是这两个催化剂均能溶于丙烯腈, 从而构成AlCl3-ZnCl2液体超强酸催化剂, 提高了催化剂的催化效率, 使双氰乙基产物收率提高.可见, 无水AlCl3是催化对位取代苯胺与丙烯腈双氰乙基加成反应的一种高效催化剂, 在丙烯腈过量较多, AlCl3的用量增加、反应温度提高的情况下, 可以获得高收率的双氰乙基加成产物; 而加入ZnCl2后构成的AlCl3-ZnCl2液体超强酸催化剂, 可使反应的收率更高.2 实验部分2.1 试剂与仪器对甲基苯胺, 对甲氧基苯胺, 对乙基苯胺, 对乙氧基苯胺, 对氯苯胺, 对硝基苯胺, 对氨基苯磺酸, 丙烯腈, AlCl3, HCl, NaAcO, ZnCl2, Na2CO3均为化学纯.北京瑞利UV 1100紫外可见分光光度计, 日本岛津LC-10AT液相色谱仪; 美国Agilent 1100 Series LC/MDS液-质联用仪; 美国Avatar-330红外光谱仪; 美国EA2400-II元素分析仪; 德国布兰克核磁共振仪(400 MHz), 北京光学仪器厂WCT-2C微机差热分析仪.液相色谱仪测试条件为: V(MeOH)∶V(H2O)=65~90∶35~10, 流速: 1~1.5 mL/min.2.2 N-氰乙基对位取代苯胺合成的典型过程在带有搅拌器、温度计和回流冷凝管的250 mL三口烧瓶中, 加入乙腈20 mL, 丙烯腈35.0 g (约0.66 mol), 无水AlCl3 1.28 g (0.0096 mol), 对甲基苯胺64.2 g (约0.60 mol), 加热至50 ℃反应4 h, 取样, 用高压液相色谱仪检测, 单氰乙基产物纯度92.5%. 然后减压蒸馏回收丙烯腈, 将瓶内物料升温至70 ℃, 倒入70 ℃水中, 加入10%稀盐酸, 搅拌, 冷却到室温, 抽滤, 再加入70 ℃水, 5% Na2CO3水溶液, 升温至70 ℃, 搅拌, 冷到室温, 过滤, 用蒸馏水洗涤至中性, 干燥, 得N-氰乙基对甲基苯胺. 经m.p., UV, IR, NMR, MS, EA分析得到确认, 并与文献基本一致[8,10].2.3 N,N-二氰乙基对位取代苯胺合成的典型过程在带有搅拌器、温度计、回流冷凝管的250 mL三口表2 对位取代苯胺的双氰乙基化反应aTable 2 Dicyanoethylation reaction of p-substituted anilines产物含量/%序号芳胺催化剂催化剂/% 丙烯腈b时间/h温度/℃单氰双氰收率/%1 p-CH3C6H4NH2 AlCl3 201∶3 16 80~85 2.6 95.9 912 p-C2H5C6H4NH2 AlCl3 201∶3 16 80~85 3.8 94.7 893 p-CH3OC6H4NH2 AlCl3 201∶4 16 75~80 3.2 95.4 904 p-C2H5OC6H4NH2 AlCl3 201∶4 16 80~85 5.1 93.5 885 p-ClC6H4NH2 AlCl3 401∶6c 16 80~85 3.1 96.5 896 p-CH3C6H4NH2 AlCl3/ZnCl2 20/10d1∶3 10 80~85 0.8 98.4 957 p-CH3OC6H4NH2 AlCl3/ZnCl2 20/10 1∶4 10 75~80 0.3 98.9 968 p-C2H5C6H4NH2 AlCl3/ZnCl2 20/10 1∶4 10 80~85 0.4 98.3 959 p-C2H5OC6H4NH2 AlCl3/ZnCl2 20/10 1∶4 10 75~80 0.4 98.5 95a对位取代苯胺用量为0.30 mol; b芳香胺与丙烯腈的物质的量比; c由于产物熔点高, 需要过量的丙烯腈作溶剂; d20/10是指AlCl3/ZnCl2的质量比.646有机化学V ol. 29, 2009烧瓶中, 加入47.7 g (约0.90 mol)的丙烯腈、6.4 g AlCl3 (0.048 mol), 保温在70 ℃使催化剂完全溶解, 得一透明液体, 再分批加入对位甲基苯胺32.1 g (约0.30 mol), 加热至80~85 ℃左右回流反应16 h, 用高压液相色谱仪检测, 双氰乙基产物纯度为95.9%. 然后减压蒸馏回收丙烯腈, 将瓶内物料升温至70~80 ℃, 倒入70~80 ℃水的烧杯中, 加入10%稀盐酸, 搅拌, 冷却到室温, 抽滤, 再加入80 ℃水, 5% Na2CO3水溶液, 升温至80 ,℃搅拌, 冷到室温, 过滤, 用蒸馏水洗涤至中性, 干燥, 得N,N-二氰乙基对甲基苯胺. 该N,N-二氰乙基化物的结构分析数据见前.2.4 物性及结构表征在本研究中, 我们获得了6种N-氰乙基对位取代苯胺和5种N,N-二氰乙基对位取代苯胺, 其中N-氰乙基对位取代苯胺已有相关文献对其物性及结构进行了表征[8,10]. 本文只对5种N,N-二氰乙基对位取代苯胺的物性及结构进行表征.N,N-二氰乙基对甲基苯胺: 灰白色固体(用乙醇重结晶后为白色针状晶体), m.p. 91~92 ℃(毛细管法), 经微机差热分析仪分析m.p. 89~92 ; b.p. 3℃14~330 ;℃UV-vis (EtOH) λmax: 211, 254, 300 nm; 1H NMR (CDCl3, 400 MHz) δ: 2.29 (s, 3H, CH3), 2.61 (t, J=6.60 Hz, 4H, CH2), 3.73 (t, J=6.64 Hz, 4H, CH2), 6.85 (d, J=8.28 Hz, 2H, C6H4), 7.13 (d, J=8.24 Hz, 2H, C6H4); IR (KBr) ν: 3023, 3013, 2962, 2915, 2856, 2245 (CN), 1619, 1522, 1478, 1459, 1409, 1359, 1324, 1179, 804, 766 cm-1; MS m/z (%): 213, 159. Anal. calcd for C13H15N3: C 73.21, H 7.09, N 19.70; found C 72.87, H 7.16, N 19.82.N,N-二氰乙基对乙基苯胺: 浅灰色或浅黄色固体, m.p. 79~81 ℃(毛细管法, 未校正, 以下同), 经微机差热分析仪分析m.p. 77~80 ℃, b.p. 293~304 ℃; UV-vis (EtOH) λmax: 212, 256, 297 nm; 1H NMR (CDCl3, 400 MHz) δ: 1.19~1.23 (m, J=7.80 Hz, 3H, CH3), 2.57~2.61 (m, J=6.52 Hz, 6H, CH2), 3.73 (t, J=6.64 Hz, 4H, CH2), 6.89 (d, J=7.76 Hz, 2H, C6H4). 7.14 (d, J=8.24 Hz, 2H, C6H4); IR (KBr) ν: 3074, 3019, 2960, 2910, 2927, 2866, 2246 (CN), 1615, 1570, 1520, 1460, 1356, 1312, 1238, 1175, 1130, 1021, 1002, 807 cm-1; MS m/z (%): 227, 173. Anal. calcd for C14H17N3: C 73.96, H 7.54, N 18.49; foundC 74.03, H 7.57, N 18.40.N,N-二氰乙基对甲氧基苯胺: 浅灰色固体或白色针状晶体, m.p. 102~103 ℃(毛细管法), 经微机差热分析仪分析m.p. 97~101 ℃; b.p. 307~325 ℃; UV-vis (EtOH) λmax: 210, 252, 311 nm; 1H NMR (CDCl3, 400 MHz) δ: 2.52 (t,J=6.72 Hz, 4H, CH2), 3.59 (t,J=6.72 Hz, 4H, CH2), 3.78 (s, 3H, CH3), 6.87 (m, J=6.72 Hz, 4H, C6H4); IR (KBr) ν: 3043, 3006, 2952, 2934, 2903, 2831, 2247 (CN), 1616, 1575, 1516, 1459, 1413, 1371, 1359, 1245, 1208, 1177, 1035, 814 cm-1; MS m/z (%): 229, 214, 175, 160. Anal. calcd for C13H15N3O: C 68.11, H 6.63, N 18.22, O 7.04; found C 68.17, H 6.63, N 18.17, O 7.03.N,N-二氰乙基对乙氧基苯胺: 浅灰色固体, m.p. 47~48 ℃(毛细管法), 经微机差热分析仪分析m.p. 45~47 ℃; b.p. 302~316 ℃; UV-vis (EtOH) λmax: 206, 250, 310 nm; 1H NMR (CDCl3,4 00 MHz) δ: 1.41 (t, J=6.80 Hz, 3H, CH3), 2.53 (t, J=6.68 Hz, 4H, CH2), 3.59 (t, J=6.68 Hz, 4H, CH2), 3.99 (q, J=6.92 Hz, 2H, CH2), 6.87 (q, J=8.88 Hz, 4H, C6H4). IR (KBr) ν: 3057, 2981, 2934, 2899, 2866, 2244 (CN), 1521, 1475, 1371, 1259, 1053, 975, 810, 790, 705 cm-1; MS m/z (%): 243, 213, 198, 169. Anal. calcd for C14H17N3O: C 69.10, H 7.05, N 17.27, O 6.58; found C 69.16, H 7.04 , N 17.15, O 6.65.N,N-二氰乙基对氯苯胺: 浅黄色或灰褐色固体. m.p. 92~93 ℃(毛细管法), 经微机差热分析仪分析m.p. 90~93 ℃; b.p. 323~340 ℃; UV-vis (EtOH) λmax: 212.5, 256, 301 nm; 1H NMR (CDCl3, 400 MHz) δ: 2.63 (m, J=6.52 Hz, 4H, CH2), 3.77 (t, J=6.56 Hz, 4H, CH2), 6.65 (d, J=8.84 Hz, 2H, C6H4), 7.27 (d, J=8.80 Hz, 2H, C6H4); IR (KBr) ν: 3054, 2971, 2957, 2915, 2250 (CN), 1597, 1570, 1503, 1459, 1422, 1397, 1373, 1290, 1227, 1204, 1174, 815, 785, 638 cm-1; MS m/z (%): 233, 198, 179, 144. Anal. calcd for C12H12N3Cl: C 61.93, H 5.21, N 18.06, Cl 14.80; found C 61.88, H 5.19, N 17.99, Cl 14.94.3 结论AlCl3在催化对位取代苯胺与丙烯腈加成反应中是一种高效催化剂. 当合成N-氰乙基对位取代苯胺时, 为防止产生双氰乙基产物, 应考虑AlCl3和丙烯腈的用量、反应温度、反应时间等因素, 具有供电子取代基团与具有吸电子取代基团的反应相比, AlCl3的用量要少, 反应温度要低, 反应时间要短些. 此外, 为抑制AlCl3强的催化活性, 同时加入NaOAc等Lewis碱是一种有效的手段, 这将有利于提高单氰乙基反应的选择性, 获得高含量、高收率的单氰乙基产物. 当合成N,N-二氰乙基对位取代苯胺时, AlCl3的用量要多, 反应温度要高, 反应时间要长, 而且加入的丙烯腈要超过化学计量. AlCl3和丙烯腈的用量、反应温度、反应时间的选择同样与对位取代基团性质有关, 具有吸电子取代基团的芳胺要求AlCl3用量多、反应温度高、反应时间长. 此外, 为提高No. 4 赵莹等:AlCl3催化对位取代苯胺与丙烯腈加成反应的研究647AlCl3的催化活性, 缩短反应时间, 加入Lewis酸ZnCl2组成的液体超强酸催化体系是一种有效的手段, 它有利于获得高含量、高收率的双氰乙基产物.References1 Yao, M.-Z.; Chen, L.-B.; Wang, J.-Y. Synthesis Principle onFine Chemicals, China Oil and Chemistry Press, Beijing, 1992, pp. 281~282 (in Chinese).(姚蒙正, 陈侣柏, 王家儒, 精细化工产品合成原理, 中国石化出版社, 北京, 1992, pp. 281~282.)2 Whitmore, F. C.; Mosher, H. S.; Adams, R. R.; Taylor, R.B.; Chapin, E.C.; Weise, C. L.; Yanko, W. J. Am. Chem.Soc. 1944, 66. 725.3 Scully, D. F. US3743668, 1973 [Chem. Abstr. 1973, 78,58104].4 Braunholtz, J. T.; Mann, F. G. J. Chem. Soc, 1953, 1817.5 Peterli, H. J.; Switzerland, B.-L. US3231601, 1966 [Chem.Abstr. 1972, 60, 2980].6 Amore, K. M.; Leadbeater, N. E.; Kristen.; Miller, T. A.;Schmink, J. R. Tetrahedron Lett. 2006, 47, 8583.7 Ross, J. M.; Farm, C.; Del, W. US3496213, 1970[Chem.Abstr. 1972, 72, 90105].8 Zhong, W.-H.; Zhang, Y.-M. Chin. J. Org.Chem. 2000, 20,747 (in Chinese).(钟为慧, 张永敏, 有机化学, 2000, 20, 749.)9 Smith, P. A. S.; Yu, T. Y. J. Am. Chem. Soc. 1952, 74,1096.10 Heininger, S. A. J. Org. Chem. 1957, 22, 1213.11 Braunholtz, J. T.; Mann, F. G. J. Chem. Soc. 1954, 651.12 Gimbert, C.; Moreno-Manas, M.; Perez, E.; Vallribera, A.Tetrahedron2007, 63, 8305.13 Zhao, Y. Fine Chem. Ind. 2001, 31, 29 (in Chinese).(赵莹, 精细化工中间体, 2001, 31, 29.)14 Zhao, Y. Chem. Eng. 2001, 83, 13 (in Chinese).(赵莹, 化学工程师, 2001, 83, 13.)15 Zhao, Y. CN 1342645, 2003 [Chem. Abstr. 2003, 138,368528g].16 Zhao, Y.; Tan, X.-Y.; Yang, Z.; Li, J.-J. Chin. J. Org. Chem.2005, 25, 1469 (in Chinese).(赵莹, 谭晓燕, 杨志, 李姣娟, 有机化学, 2005, 25, 1469.)17 Sun, X.-W.; Zhao, S.-Q. Acta Chim. Sinica2008, 66, 471(in Chinese).(孙学文, 赵锁奇, 化学学报, 2008, 66, 471.)18 Jiang, W.-W. Fine. Chem. 1997, 14, 46 (in Chinese).(蒋文伟, 精细化工, 1997, 14, 46.)(Y0803213 Zhao, X.; Zheng, G.)。
有机胺
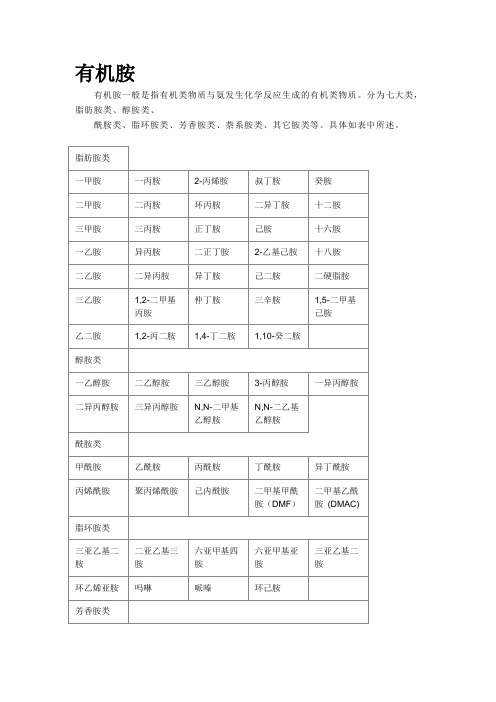
有机胺有机胺一般是指有机类物质与氨发生化学反应生成的有机类物质。
分为七大类,脂肪胺类、醇胺类、酰胺类、脂环胺类、芳香胺类、萘系胺类、其它胺类等。
具体如表中所述。
聚乙烯亚胺羟胺絮凝剂理论基础是;“聚并”理论,絮凝剂主要是带有正电(负)性的基团中和一些水中带有负(正)电性难于分离的一些粒子或者叫颗粒,降低其电势,使其处于不稳定状态,并利用其聚合性质使得这些颗粒,集中,并通过物理或者化学方法分离出来。
一般为达到这种目的而使用的药剂,称之为絮凝剂。
絮凝剂主要应用于给水各污水处理领域。
絮凝剂按照其化学成分总体可分为无机絮凝剂和有机絮凝剂两类。
其中无机絮凝剂又包括无机凝聚剂和无机高分子絮凝剂;有机絮凝剂又包括合成有机高分子絮凝剂、天然有机高分子絮凝剂和微生物絮凝剂。
[编辑本段]无机絮凝剂按其分子量的大小可分为低分子絮凝剂和高分子絮凝剂两大类。
低分子絮凝剂价格低、货源充足、但因其用量大、残渣多、效果差,故无机絮凝剂的发展已经基本上完成了低分子向高分子的转变。
现常用的无机高分子絮凝剂有聚合铝类絮凝剂、聚合铁类絮凝剂和活性硅酸类絮凝剂以及复合絮凝剂四大类。
(1)聚合铝类絮凝剂(如聚合氯化铝,硫酸铝等)聚合铝水解产生高价离子,形成各种类型的羟基多核络合物。
它们通过羰基式桥联作用,处于亚稳定状态。
而OH-与Al3+的比值[2](一般称盐基度或碱基度)对絮凝效果影响很大。
通常盐基度越高,絮凝效果越强,但过高则本身易生成难溶的氢氧化铝沉淀,导致絮凝效果降低。
研究表明,盐基度在75%-85%时最佳,此时絮凝体产生快,颗粒大而重,沉淀性能好。
聚合铝具有投药量少、沉降速度快、颗粒密实、除浊、除色效果明显等特点。
在工业水处理中得到广泛的应用[3]。
值得注意的是铝,尤其是活性铝,毒性较大,同时聚合铝制备方法不完善,致使较多水解铝的微细颗粒存在于溶液中,这在一定程度上限制了聚合铝的使用。
通过改善混凝反应条件,延长慢速混凝时间,能有效降低水中铝的含量。
有机胺
有机胺有机胺一般是指有机类物质与氨发生化学反应生成的有机类物质。
分为七大类,脂肪胺类、醇胺类、酰胺类、脂环胺类、芳香胺类、萘系胺类、其它胺类等。
具体如表中所述。
聚乙烯亚胺羟胺絮凝剂理论基础是;“聚并”理论,絮凝剂主要是带有正电(负)性的基团中和一些水中带有负(正)电性难于分离的一些粒子或者叫颗粒,降低其电势,使其处于不稳定状态,并利用其聚合性质使得这些颗粒,集中,并通过物理或者化学方法分离出来。
一般为达到这种目的而使用的药剂,称之为絮凝剂。
絮凝剂主要应用于给水各污水处理领域。
絮凝剂按照其化学成分总体可分为无机絮凝剂和有机絮凝剂两类。
其中无机絮凝剂又包括无机凝聚剂和无机高分子絮凝剂;有机絮凝剂又包括合成有机高分子絮凝剂、天然有机高分子絮凝剂和微生物絮凝剂。
[编辑本段]无机絮凝剂按其分子量的大小可分为低分子絮凝剂和高分子絮凝剂两大类。
低分子絮凝剂价格低、货源充足、但因其用量大、残渣多、效果差,故无机絮凝剂的发展已经基本上完成了低分子向高分子的转变。
现常用的无机高分子絮凝剂有聚合铝类絮凝剂、聚合铁类絮凝剂和活性硅酸类絮凝剂以及复合絮凝剂四大类。
(1)聚合铝类絮凝剂(如聚合氯化铝,硫酸铝等)聚合铝水解产生高价离子,形成各种类型的羟基多核络合物。
它们通过羰基式桥联作用,处于亚稳定状态。
而OH-与Al3+的比值[2](一般称盐基度或碱基度)对絮凝效果影响很大。
通常盐基度越高,絮凝效果越强,但过高则本身易生成难溶的氢氧化铝沉淀,导致絮凝效果降低。
研究表明,盐基度在75%-85%时最佳,此时絮凝体产生快,颗粒大而重,沉淀性能好。
聚合铝具有投药量少、沉降速度快、颗粒密实、除浊、除色效果明显等特点。
在工业水处理中得到广泛的应用[3]。
值得注意的是铝,尤其是活性铝,毒性较大,同时聚合铝制备方法不完善,致使较多水解铝的微细颗粒存在于溶液中,这在一定程度上限制了聚合铝的使用。
通过改善混凝反应条件,延长慢速混凝时间,能有效降低水中铝的含量。
EN 14362-1 2012标准学习
序号
化学文摘号
索引号
从纺织品中选出有色测试样品,测试样品依据分散染料萃取的方法和(或) 其他种类染料直接还原的方法测试。 根据测试样品的情况,使用2种方法中的1种或结合2种方法。 使用其他种类染料的处理方法时,测试样品在密闭容器中70℃柠檬酸盐缓冲 溶液(PH=6)里使用连二亚硫酸钠还原处理。 当萃取分散染料时,使用氯苯回流萃取。萃取液经浓缩后用甲醇转移到反应 器中,后面在70℃柠檬酸盐缓冲溶液(PH=6)中使用连二亚硫酸钠还原. 如果纺织样品在使用氯苯萃取后没有完全褪色,把样品和分散染料中的甲醇 溶液加入反应器中一起还原。 还原后,过程中释放的任何胺通过。 本方法描述了两种提取方法:一种是使用硅藻土柱进行萃取,再对叔丁基甲 基醚萃取液进行浓缩,上机检测。另一种是不使用硅藻土柱进行液液萃取的 定性筛选方法。 如果任何一种胺被检测出来,需要使用1种或多种选择性的方法进行确认。
3)如果达不到测试质量(1g),则需取多份相同 部件;
◦ 如果材料的总质量低于0.5g,这个材料定义为次要的成分
(附录C备注2中指出:样品量太少,所得结果具有很大的不 确定性。).
◦ 质量低于0.2g的材料可忽略不计。 ◦ 印花连同底布一起称重,不需分离
2.6.2 单一纤维成分
标准中第10部分 结果评价中明确指出:当任一种胺使用 单点(15mg/L)定量的结果超过5mg/kg,就应建立一 个多点的校正曲线进行定量。
若萃取后完全褪色,则摈弃测试样品。 若未完全褪色,则用正戊烷或叔丁基甲基醚冲洗样品并使其干 燥后,放入反应器中,合并甲醇溶解的分散染料溶液(2mL) 一起还原。
2.7.2 其他染料染色的纺织品
有机化学习题化学
CH 3CH C(CH 3)2(CH 3)2CHCH 2CH CHCHCH 2CH 33C C HCl BrCH 2CH 3C C CH 2CH 2CH 3CH 3H CH(CH 3)2CH 3CH 2C(CH 3)2CCHC CH 2CH 3CH 2CH 3+ HCl CF 3CH CH 2+ HCl(CH 3)2CCH 2+ Br 22单元练习21. 用系统命名法命名下列化合物(1) (2) (3)(7) (8)2. 写出下列化合物结构式或构型式,如其名称与系统命名原则不符,请予更正: (1) 6,6–二甲基–4–乙基壬烷 (2) 2,4–二甲基-3–异丙基庚烷 (3) 2–异丙基-4–甲基戊烷 (4) 2,3–二甲基–2–乙基丁烷 (5) 1–甲基–3–环丙基环戊烷 (6) 反-1-溴-4-氯环己烷(优势构象) (7) 4–甲基螺[2.4]庚烷 (8) 1,2,4–三甲基二环[4.4.0]癸烷3. 写出庚烷的各种构造异构体,并用系统命名法命名4. 写出下列化合物的可能结构式:(1) 由一个异丙基和一个异丁基组成的烷烃;(2) 只含一个叔丁基和一个甲基,分子量为140的单环烷烃的顺式异构体; (3) 含一个伯碳、四个仲碳和一个叔碳的环烷烃; (4) 分子式C 6H 14,只含伯碳和叔碳。
5. 不查表比较下列各化合物沸点的高低: (1) (2)(3) (4) 6. 写出异戊烷所有一氯代产物结构式,并将其按氢原子反应活性排列。
7. 写出下列化合物的优势构象:(1) (2)(3) 顺–1–甲基–3–乙基环己烷 (4) 反–1–甲基–3–乙基环己烷 8. 用简单化学方法区别下列各组化合物:(1) 环丙烷和丙烷 ;(2) 1–甲基–2–乙基环丙烷和甲基环戊烷 9. 推导结构式:化合物A 、B 、C 是互为同分异构体的烷烃,分子量均为72。
A 中仅含有伯碳和季碳,B 中含有伯、仲、叔碳,C 中含有伯、仲碳原子。
第四章 化学合成工艺的设计与选择01
发展迅速的喹诺酮类抗菌药的基本骨架相似,合成以 多取代苯胺为原料,构建吡酮酸环。 构建方法是在诺氟沙星(norfloxacin)和环丙沙星 (ciprofloxacin)等早期品种的合成经验基础上发展而来 的,是典型的模拟类推法的应用实例,如氟罗沙星 (fleroxacin)和加替沙星(gatifloxacin)的合成工艺路 线。
一、 类型反应法 将已知的经典合成方法用于产品或中间体合成路线设 计的方法,称为类型反应法。 类型反应法主要应用于那些没有现成的合成方法可供 参考或已知的合成方法不够合理的场合。 研究内容包括分子骨架的形成、官能团的引入与转换 以及敏感基团的保护与脱保护等。
药物克霉唑为三芳甲基咪唑类化合物,由反合成 分析,可将其切断为合成等价物2-氯苯基二苯基氯甲 烷和咪唑。
第四章 化学合成工艺的设计与选择
一个产品常常可以由多种工艺路线合成。 化学工业中,选择先进、可靠、经济、安全的化合 物合成方法是实现化学合成产业化的第一命题。 工艺路线:一个化学合成产品往往可通过多种不同 的合成途径制备,通常将具有工业生产价值的合成途径 称为该产品的工艺路线。 在化学合成产品的工艺研究中,首先是工艺路线的 设计和选择,以确定一条经济而有效的生产工艺路线。
2专利即将到期的产品化工产品的专利到期后其它企业便可以仿制化工产品的价格将大幅度下降成本低价格廉的生产企业将在市场上具有更强的竞争力设计选择合理的工艺路线显得尤为重3产量大应用广泛的产品某些老产品社会需求量大应用面广如能设计选择更加合理的工艺路线简化操作程序提高产品质量降低生产成本减少环境污染可为企业带来极大的经济效益和良好的社会效益
设计与选择合成工艺的一般方法: ①收集(类似)化合物的文献合成方法,要求尽量全,不要遗 漏。 ②结合理论与实际,研究论证各方法的合理性(理论上是否 合理等)、可行性(原料是否易得,反应条件是否苛刻等)和 经济性(是否有利可图)等。同时,尽量考虑采用新技术新工 艺(如生物技术、催化技术)的可能性。 ③给出总结报告,写出研究或生产预案。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
名称 摩 尔质 量 投 抖 量 摩 尔 效 摩 尔 比 硝 基 物
= 甲 苯
11 有 机 溶 剂 介质 中 的硝 化 .
试 验采用 了苯 、 甲苯 、 氯苯 、 甲烷 、 氯 二氯 乙烷 、 乙醇等溶 剂 , 中以 1 一二氯 乙烷效果最好 , 其 . 2 且溶 剂的使 用量直接影 响
产品的质量和收率。 在二氯甲烷溶剂 中产品的熔点 1 0 1 1 , 4 ~4 ℃ 收率9 — 8 其投料量和操作方法 如表 1 2 9 %, :
表 1 投料 量
名 称 M 投料 量 摩尔数 ( o ) 摩 尔比 o t1 03 .0
0. 53
鲁注 含量 9
2 5 l 7
在装有搅拌 器、 温度计 、 冷却 器的 2 0 5 mL四 口烧瓶 中加 入 3 mL .一二氯乙烷 , 0 1 2 搅拌下加入 5 .g 28 氯化物使其溶解 , 温 升
至 6 ℃ , 3 mi 0 在 0 n内滴 加 4 %的 HN 7 (7 6 % HNO 7 O 5 mL3 mL 5
滤、 干燥得灰 白色产品。
22 加 氢 还 原 法 .
加氢还原法工艺较铁粉还原法复杂 , 实施起来有一定难度 , 但该方法对 于提高产品质量和收率 , 以及保护环境都较铁粉还 原法优越 。 该法得到的产品外观为 白色 , 含量在 9 %以上 , 5 收率
可达 8 %以上 。其投料量和操作方法如下 : 5 操作方法 :
硝化
7℃, 0 搅拌下在 3 mi 0 n内滴加 5 .g 甲氧基氯苯 , 7 ℃下 28 二 在 0 保温反应 2 , h 过滤 , 将滤饼洗至 中性 , 用水 抽干 , 干燥 , 得产品。
表 2 投料量
酸 ,溶剂 或水 F 或 Nhomakorabea褡 剂介质 e
2 还 原
1 硝化
25 . 一二甲氧基氯苯 ,环上有 两个致活 的 一 C 0 H 和一个 致 钝 的 一 l酚醚类 硝化一般采用 硝酸硝化 , C, 故本 硝化亦 采用 该
维普资讯
20 0 7年 6月
广 西 轻 工 业
G A G I O R A FL H D SR U N X U N L O I TI U T Y J G N
∞
第 6期( 总第 1 3 ) 0 期
化 工 与 材 料
由 25 .一二 甲氧基氯苯合成 4 一氯 一 .一二甲氧基苯胺研 究 25
李振年 , 高平
( 定 平安 安全评 价 有 限公 司, 保 河北 保 定 0 0 0 7 0 ) 1
【 摘 要】本文 对由2 一 氧基氯苯合成4 氯一 . 二甲 . 二甲 5 一 2 一 氧基苯胺的过 5 程进行了实 研究, 验、 通过对比, 确定了 投料配
比、 助 催 化 剂 、 化 和 还 原 工 艺路 线 。 辅 硝
为了简化工艺 , 降低原料消耗 , 在原有 工艺 基础上 又探索 了水介 质中硝化的方法 , 对加料方式 、 温度 、 硝酸浓度 、 量及 用
反应时间等条件进行 了研究 实验 , 取得 了最佳反应条 件 , 实验
结果为 : 品熔点 1 95C 1 05C, 产 3 . ̄~ 4 . ̄ 收率 9 %以上 。其投料量 6
方法 。
该硝基物的还原 , 可采 用铁粉和加氢还原两种方 法。
21 铁 粉还 原 法 .
铁粉还原 法工艺简单 、 较易实施 、 成本低 , 品外 观较差 , 产 但也能满足要求。铁粉还原法得到 的产 品外 观浅灰 白色 , 含量
大于 9 %, 0 收率高于 8 %。其投料量和操作方法如下 : 0
+ 0 H: )滴完后升温至 7  ̄搅 拌反应 2 , 滤 , 2 mL 0 , 0C h抽 滤饼用水 洗至中性, 再用约 3 m : 0 L11的乙醇 ~水 , 冲洗滤饼 , 干 , 抽 干燥
得产品。 1 水 介质 中 的硝 化 . 2
过滤 , 收集滤液 , 自然冷却 , 又析 出少量产品。 合并 两次滤液 , 过
和操作方法如下 : 在装有搅拌 器、 温度计 、 冷却 器的 2 0 5 mL四口烧 瓶中加入
l6 0
2g 0
6 mL 0
O9 . 0
03 6
l
4
铁 粉 醋酸
NH CI
5 6 6 0
5 35
2g 0 2 mL
05 .g
H 2 0
l 8
2 mL 0
备 注 含量 9 % 8
d . 3l =1 9
二甲氧 基氯 苯 1 25 5 .g 7. 28 1 一 二氯 乙烷 9 . 2 9
【 关键词 】4 氯 一. 二甲 一 2一 5 氧基苯胺; — P C催化剂; t 硝化; 还原 【 中图分类号 】 R 1 97 【 文献标识码 】 A 【 文章编号 】10— 63 07 6 03 — 2 0327( 0) — 0 1 0 2 0
4 一氯 一 .一二 甲氧基苯胺是染料 、 药等重要 的中间体 , 25 农 该产品的合成包括两步化学 反应 : 1 .一二 甲氧基氯苯 的硝 ( )25 化 ;2 ( )所得硝基物 的还原 , 其反应式为 :
N 1 H C 搅拌 加热至 回流 ( 9 "左右 )保 持 3 mi , 约 7C , 0 n 加入 1 g 0 硝 基物 , 搅拌均匀后加 入另一半铁 粉( 0 )搅拌 回流 3 mi, 1g , 0 n 再加入另外 1 g 硝基物 , 0 搅拌加热 3 mi , 0 n 趁热过滤 , 收集滤液 自然冷却 , 析出产品。 铁泥再加入 4 ~ 0 0 5 mL二甲苯 回流 3 mi, 0 n
6 %HNOt 5 H2 0 6 3 l 8
l
177 .
3 mL 0
3 7mL 2 0mL
操作 : 在装有 温度 计、 冷却器及搅拌器 的 2 0 5 mL四口烧瓶
中 ,加入 6 mL二甲苯 ,0 铁粉 , mL醋 酸 、0 0 1g 2 2 mL水和 05 .g