基因工程 第8章 PCR技术
基因组学杨金水电子版 基因工程 电子版

基因组学杨金水电子版基因工程电子版导读:就爱阅读网友为您分享以下“基因工程电子版”的资讯,希望对您有所帮助,感谢您对的支持! 作者:吴乃虎出版社:高等教育出版社第一章基因工程概述第一节基因操作与基因工程一、基因操作与基因工程的关系二、基因工程的诞生与发展第二节基因工程是生物科学发展的必然产物一、基因是基因重组的物质基础二、DNA的结构和功能三、基因操作技术的发展促进基因工程的诞生和发展四、基因工程的内容第三节基因的结构——基因操作的理论基础一、基因的结构组成对基因操作的影响二、基因克隆的通用策略第一篇基因操作原理第二章分子克隆工具酶第一节限制性内切酶一、限制与修饰二、限制酶识别的序列三、限制酶产生的末端四、DNA末端长度对限制酶切割的影响五、位点偏爱六、酶切反应条件七、星星活性八、单链DNA的切割九、酶切位点的引入十、影响酶活性的因素十一、酶切位点在基因组中分布的不均一性第二节甲基化酶一、甲基化酶的种类二、依赖于甲基化的限制系统三、甲基化对限制酶切的影响第三节DNA聚合酶一、大肠杆菌DNA聚合酶二、KIenow DNA聚合酶三、T4噬菌体DNA聚合酶四、T7噬菌体DNA聚合酶五、耐热DNA聚合酶六、反转录酶七、末端转移酶第四节其他分子克隆工具酶一、依赖于DNA的RNA聚合酶二、连接酶三、T4多核苷酸激酶四、碱性磷酸酶五、核酸酶六、核酸酶抑制剂七、琼脂糖酶八、DNA结合蛋白九、其他酶第三章分子克隆载体第一节质粒载体一、质粒的基本特性二、标记基因三、质粒载体的种类第二节λ噬菌体载体一、λ噬菌体的分子生物学二、λ噬菌体载体的选择标记……第四章人工染色体载体第五章表达载体第六章基因操作中大分子的分离和分析第七章基因芯片技术第八章PCR技术及其应用第九章DNA序列分析第十章DNA诱变第十一章DNA文库的构建和目的基因的筛选第十二章基因组研究技术第二篇基因工程应用第十三章植物基因工程第十四章动物基因工程第十五章酵母基因工程第十六章细菌基因工程第十七章病毒基因工程第十八章医药基因工程第十九章基因工程产品的安全及其管理第一章基因工程概述第一节基因操作与基因工程一、基因操作与基因工程的关系基因操作(gene manipulation):指对基因进行分离、分析、改造、检测、表达、重组和转移等操作的总称。
高中生物高考考点80 PCR技术-备战2022年高考生物考点一遍过
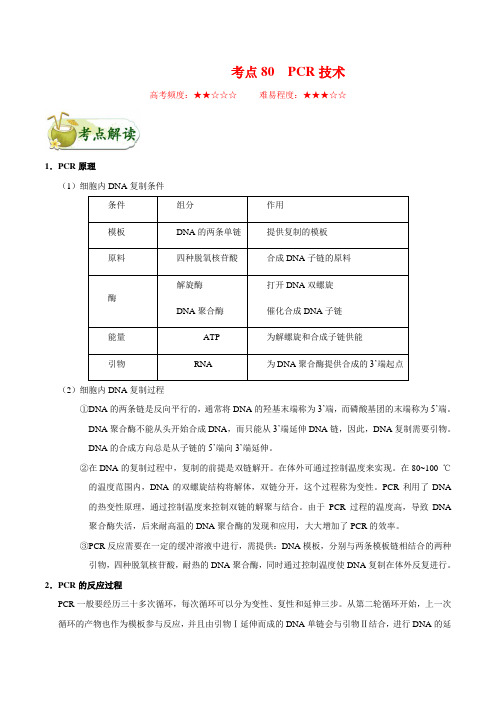
考点80 PCR技术高考频度:★★☆☆☆难易程度:★★★☆☆1.PCR原理(1)细胞内DNA复制条件条件组分作用模板DNA的两条单链提供复制的模板原料四种脱氧核苷酸合成DNA子链的原料酶解旋酶DNA聚合酶打开DNA双螺旋催化合成DNA子链能量ATP 为解螺旋和合成子链供能引物RNA 为DNA聚合酶提供合成的3’端起点(2)细胞内DNA复制过程①DNA的两条链是反向平行的,通常将DNA的羟基末端称为3’端,而磷酸基团的末端称为5’端。
DNA聚合酶不能从头开始合成DNA,而只能从3’端延伸DNA链,因此,DNA复制需要引物。
DNA的合成方向总是从子链的5’端向3’端延伸。
②在DNA的复制过程中,复制的前提是双链解开。
在体外可通过控制温度来实现。
在80~100 ℃的温度范围内,DNA的双螺旋结构将解体,双链分开,这个过程称为变性。
PCR利用了DNA的热变性原理,通过控制温度来控制双链的解聚与结合。
由于PCR过程的温度高,导致DNA聚合酶失活,后来耐高温的DNA聚合酶的发现和应用,大大增加了PCR的效率。
③PCR反应需要在一定的缓冲溶液中进行,需提供:DNA模板,分别与两条模板链相结合的两种引物,四种脱氧核苷酸,耐热的DNA聚合酶,同时通过控制温度使DNA复制在体外反复进行。
2.PCR的反应过程PCR一般要经历三十多次循环,每次循环可以分为变性、复性和延伸三步。
从第二轮循环开始,上一次循环的产物也作为模板参与反应,并且由引物Ⅰ延伸而成的DNA单链会与引物Ⅱ结合,进行DNA的延伸,这样,DNA聚合酶只能特异地复制处于两个引物之间的DNA序列,使这段固定长度的序列成指数扩增。
3.实验操作(1)PCR反应体系:缓冲液、DNA模板,四种脱氧核苷酸原料、热稳定的DNA聚合酶、两种RNA引物、水。
(2)实验操作步骤①按照PCR反应体系配方配制反应液;②将PCR反应体系50μL用微量移液器转移到微量离心管(0.5 mL)中;③将微量离心管放到PCR仪中;④设置PCR仪的工作参数;⑤DNA在PCR仪中大量扩增。
大学基因工程复习归纳重点复习资料

基因工程复习归纳第一章绪论1.基因工程的定义:是指按照人们的愿望,经过严密的设计,将一种或多种生物体(供体)的基因与载体在体外进行拼接重组,然后转入另一种生物体(受体/宿主)内,使之按照人们的意愿稳定遗传、并表达出新的性状的技术。
2.基因工程概念的发展:遗传工程→DNA重组技术→分子/基因克隆(Molecular/Gene→基因工程→基因操作。
应用领域以“基因工程”、“DNA重组”为主基因工程基因工程的历史性事件1973:Boyer和Cohen建立DNA重组技术1978:Genetech公司在大肠杆菌中表达出胰岛素1982:世界上第一个基因工程药物重组人胰岛素上市1988:PCR技术诞生1989:我国第一个基因工程药物rhIFNα1b上市2003: 世界上第一个基因治疗药物重组腺病毒-p53上市3.基因工程的三大关键元件基因(供体):外源基因、目的基因载体:能将外源基因带入受体细胞,并能稳定遗传的DNA分子(克隆载体、表达载体)。
宿主(受体):,能摄取外源DNA、并能使其稳定维持的细胞(组织、器官或个体)。
4.基因工程的基本步骤(切、接、转、增、检(大肠杆菌是中心角色)(1)目的基因的获取:从复杂的生物基因组中,经过酶切消化或PCR扩增等步骤,分离出带有目的基因的DNA片断。
(2)重组体的制备:将目的基因的DNA片断插入到能自我复制并带有选择性标记(抗菌素抗性)的载体分子上。
(3)重组体的转化:将重组体(载体)转入适当的受体细胞中。
(4)克隆鉴定:挑选转化成功的细胞克隆(含有目的基因)。
(5)目的基因表达:使导入寄主细胞的目的基因表达出我们所需要的基因产物。
第二章 DNA重组克隆的单元操作一、用于核酸操作的工具酶1.限制性核酸内切酶(主要存在于原核细菌中,帮助细菌限制外来DNA的入侵)。
限制性核酸内切酶的功能与类型其中II型限制性核酸内切酶:切割位点专一,适于DNA重组,是DNA重组中最常用工具酶。
基因工程中PCR技术常考知识点的归纳与拓展

基因工程中PCR技术常考知识点的归纳与拓展作者:***来源:《中学教学参考·理科版》2024年第05期[摘要]PCR技术是基因工程中获取和扩增目的基因的常用方法之一,其中常考查到的知识点有与引物相关的问题、酶切片段分析及变性、复性、延伸等环节的相关分析等。
文章结合典型考题对这几个问题进行归纳分析。
[关键词]基因工程;PCR技术;常考知识点[中图分类号] G633.91 [文献标识码] A [文章编号] 1674-6058(2024)14-0080-04【名师简介】曾凡洪,中学高级教师,武汉市学科带头人,曾担任高中生物奥赛主教练。
先后在《生物学教学》《中学生物教学》《中学教学参考》《高中生学习》等报纸杂志上发表文章50余篇,并参与多本著作的编写。
聚合酶链式反应(PCR)是一种用于扩增特定DNA片段的分子生物学技术,它可看作是生物体外的特殊DNA复制,其最大特点是能将微量的DNA大幅增加。
因此,无论是化石中的古生物、历史人物的残骸,还是几十年前凶杀案中凶手所遗留的毛发、皮肤或血液,只要能分离出一丁点的DNA,就能用PCR加以放大后进行比对,这也是“微量证据”的威力之所在。
PCR的理论依据是DNA复制,一般的中学实验室很难具备相应的实验设备和操作条件,因此对于这块知识,基本上是以理论讲解为主。
目前的一些考题涉及的PCR知识已经比较具体和深入了,具有一定的难度。
本文结合典型考题对基因工程中PCR技术常考知识点进行归纳分析。
一、与引物相关的问题引物是一小段单链DNA或RNA,是DNA复制的起始点。
在引物的3'—OH上,核苷酸以酯键形式进行加成,因此引物的3'—OH必须是游离的。
引物分为两种:存在于自然生物的DNA复制引物(RNA引物)和PCR中人工合成的引物(通常为DNA引物)。
一般所说的引物是指DNA引物。
PCR中的引物是人工合成的两段寡脱氧核苷酸序列。
(一)设计引物的基本要求根据DNA复制原理和引物的作用,在设计引物时,要注意以下几个基本问题。
简述PCR扩增的原理和步骤

简述PCR扩增的原理和步骤一、背景简介PCR(Polymerase Chain Reaction)即聚合酶连锁反应,是一种在分子生物学中广泛应用的技术,其原理是通过循环性的DNA复制过程,在体外大量扩增特定DNA片段。
PCR扩增技术的发展极大地推动了基因工程、医学诊断和遗传学研究等领域的进展。
二、PCR扩增的原理PCR扩增的原理是通过特定的酶和引物使靶DNA序列在体外进行酶催化反应,从而实现目标DNA的扩增。
主要涉及以下三个步骤:变性、退火和扩增。
•变性(Denaturation):将待扩增的DNA样本加热至94-98摄氏度,使DNA双链解开成两条单链。
这个步骤中,高温会导致DNA的氢键断裂,使双链DNA解旋成单链DNA。
•退火(Annealing):将温度降低至50-65摄氏度,使两条单链DNA的引物与靶DNA序列互相结合。
引物是特异性碱基序列,与待扩增的DNA序列的两端互补配对。
•扩增(Extension):将温度升高至72摄氏度,引入热稳定性DNA聚合酶。
该酶会沿着引物朝着3’方向合成新的DNA链。
此步骤中,引物会向前、向后识别并结合到两条单链DNA的特异DNA酶切位点,然后在这些位置上进行DNA链合成。
这样,每个循环会产生两个新的DNA分子。
三、PCR扩增的步骤1.样本处理:从待扩增的样本中提取出DNA,并进行纯化。
这一步骤是为了去除样本中的RNA、蛋白质等物质,以保证反应的准确性和特异性。
2.引物设计:根据目标DNA的序列,设计引物。
引物通常由20-30个核苷酸组成,需要具有与目标DNA序列的两端互补的碱基序列。
3.PCR反应:在PCR反应管中加入待扩增的DNA样本、引物、核苷酸和DNA聚合酶等反应物,并按照特定的温度和时间进行反应。
一般情况下,PCR反应包括变性、退火和扩增三个步骤,其循环次数取决于所需扩增的DNA的数量。
4.扩增产物检测:通过凝胶电泳或其他相关技术,检测PCR扩增产物的大小和纯度。
PCR技术及应用

2、引物的设计: 引物包括两条,即5/端引物和3/端引物。 引物决定PCR扩增产物的特异性和长度
引物设计的基本要求: (1)引物长度: 15~30 nucleotides (2)碱基的分布尽可能随机,避免出现嘌 呤或嘧啶碱基的堆积现象。 No GGG or CCC at 3’-end (G+C) 45%~ 55% Tm=4(G+C)+2(A+T)
(3)dNTP 能与Mg2+结合,使游离的Mg2+ 浓度降低
5)镁离子浓度 TaqDNA聚合酶的激活剂 Mg2+对PCR扩增的特异性和产量有显著 的影响;在一般的 PCR反应中,各种NTP浓 度为200umol/L时,Mg2+浓度为1.5~2.0 mmol/L为宜; Mg2+浓度过高,反应特异性降低,出现非 特异扩增, 浓度过低会降低,Taq DNA聚合酶的活性 不够,使反应产物减少。
PCR技术简史
• DNA的复制 • 核酸体外扩增的设想 • 聚合酶链反应的发明
• 1985年,美国PE-Cetus公司的Mullis等人发明了 聚合酶链反应(PCR)
• 基本原理是在试管中模拟细胞内的DNA复制
• 最初采用E-coli DNA聚合酶进行PCR,由于该酶 不耐热,使这一过程耗时,费力,且易出错 • 耐热DNA聚合酶的应用使得PCR能高效率的进行, 随后PE-Cetus公司推出了第一台PCR自动化热循 环仪 • 1993年,Mullis等因此项技术获诺贝尔化学奖
含有解螺旋酶、DnaC蛋白、引物酶和DNA 复制起始区域的复合结构称为引发体。
复制的起始
3
5 3
引物
5
引物是由引物酶催化合成的短链RNA分子。
复 制 过 程 简 图
PCR技术及应用

聚合酶链式反应
——PCR技术
PCR (Polymerase chain reaction) 是用一对寡聚DNA 作为引物,通过加温变性-退火-延伸(DNA合成) 这一周期的多次循环,使目的DNA片段得到扩增。 由于这种扩增产物是以指数形式积累的,经25~30 个循环后,扩增倍数可达106。
中国的状况 1991年出现三个水浴锅+机械手的原始PCR仪(华美,复日等) 现在以半导体(Peltier板)制冷式为主(杭州博日,上海天呈, 厦门安普 利等)
耐高温DNA聚合酶的种类
Taq
DNA聚合酶 Tth DNA聚合酶 Pfu DNA聚合酶
Tfl
(Thermus aquaticus,Cetus/Roche)
目前,TALEN 已经成功应用于酵母、 哺乳动
物和植物的位点特异性基因打靶,与锌指核酸 酶系统相比有较大的应用优势,但仍然有些问 题需要解决,例如:脱靶效应、TALEN 与基因 组进行特异结合与染色体位置及邻近序列有关 等。
3. CRISPR /Cas9 基因组编辑技术
CRISPR (Clustered regularly interspaced short
充满梦想却幻灭的ZFN:
难以完全找到匹配的3连子锌指; • Off-target 严重 • 美国加州大学Dave Segal教授说: “ With zinc fingers, we have to let the DNA tell us where we target”。
•
2. TALEN 基因组编辑技术
PCR不只是一个方法改进
Mullis的上司有句“名言”,“我们
要扩增这么多DNA样品有什么用”。 到了1991,Cetus公司以3亿美元的 转让费将PCR相关专利转让给瑞士 Hoffman LaRoche公司,并在此后 的经营中又获得了数亿美元的分红。 Mullis于1993年获得诺贝尔化学奖, PCR和DNA重组技术一样意义深远。
基因工程中目的基因的检测pcr法

基因工程中目的基因的检测pcr法基因工程中目的基因的检测PCR法在现代生物技术领域,基因工程扮演着至关重要的角色。
它涉及到对生物体的基因进行精确的修改和调控,以实现特定的科学或商业目标。
在基因工程的过程中,检测目的基因的存在和表达是至关重要的一步。
聚合酶链反应(PCR)作为一种高效、灵敏的基因检测技术,已被广泛应用于这一领域。
PCR技术的基本原理是利用DNA聚合酶在特定条件下,对目标DNA 序列进行指数级扩增。
这一过程涉及到几个关键步骤:变性、退火和延伸。
通过高温将双链DNA变性为单链,然后通过降温使引物与目标序列特异性结合,在适宜的温度下,DNA聚合酶沿着模板链合成新的DNA链。
通过这一系列的循环,目标DNA序列得以在短时间内大量复制,从而便于检测和分析。
在基因工程中,PCR技术被用于多种目的。
它可以用于验证目的基因是否已成功导入宿主细胞。
通过设计特异性的引物,可以扩增出导入的基因片段,并通过凝胶电泳等方法进行检测。
PCR还可以用于检测目的基因的表达水平。
通过逆转录PCR(RTPCR)技术,可以将RNA转录为cDNA,然后通过PCR扩增,从而间接检测基因的表达量。
为了确保PCR检测的准确性和可靠性,必须严格控制实验条件。
引物的设计至关重要。
引物需要与目标序列完全匹配,并且具有适当的长度和GC含量,以确保高效性和特异性。
实验中的温度控制、试剂浓度和循环次数等参数也需要精确调控。
任何微小的偏差都可能导致结果的误判。
在应用PCR技术进行基因检测时,还需要考虑到可能存在的假阳性或假阴性结果。
假阳性通常是由于交叉污染或非特异性扩增引起的,而假阴性可能是由于模板量不足或扩增条件不适宜导致的。
因此,在实验设计中,应采取适当的对照和重复实验,以验证结果的可靠性。
随着PCR技术的不断发展,出现了许多基于PCR的衍生技术,如定量PCR(qPCR)、多重PCR和高通量PCR等。
这些技术不仅提高了检测的灵敏度和特异性,还实现了对多个基因的同时检测,大大提高了实验效率。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
每 循 环 的 效 率 (%)
错误率(错误/ 拷入碱基对)
PCR 引 起 的 突 变
需 要 的 循 环数
60 56 90 7.0 36 88 80
7×10 -6 5×10 4.4×10-5 4.5×10-5 7.2×10-5 2×10 -4 1.3×10
-4
-7
0.3 2 13 16 25 56 41
Tm=(G+C)×4+(A+T)×2 for short primers × ×
Operon 公司
③ 避免二级结构的形成 二聚体 二级结构 ④ G/C和A/T分布尽量均匀,一般G+C%45-55% oligo dT /oligo dC 除外, ⑤ 其它,如5’末端,限制酶位点的长度 ⑥ rondom primers 随机引物 6nt 10-12nt
3.Time .
Denature 30” 若G+C高可长,若为Cell可长 Renature 30” (20”-40”) Extention 1min >>1kb 55℃ 24nt/sec 37℃ 1.5nt/sec 72℃ 500bp--- 20sec, 1.2kb--- 40sec if secondary structure, longer time
∆ Basic principle:DNA thynthesing template/primer/DNA poly ∆ Application: themal-resistance DNA polymerase
DNA amplification Principle
template primer DNA polymerase 5' 3' 3' 5'
30 32 22 26 45 22 24
德维克斯勒, 科学出版社 C.W.迪芬巴赫 /G.S德维克斯勒,1998 迪芬巴赫 德维克斯勒 C.W.Dieflenbach/G.S.Dueksler
诱变) (5)PCR-mediated mutaginesis(诱变 ) 诱变
Chapter 8
PCR technology
PCR
polymerase chain reaction
Another cloning method
Selective amplification of a DNA sequence. In some situation, it essentially replaces traditional cloning methodology.
(2) Tth DNA polymerase
Thermus thermophilus HB8, 94kDa, Mg2+: 5’ ---3’合成DNA Mn2+: 以RNA为模板合成cDNA 可解决RNA反转录过程中形成的二级结构 ·反转录: primer extension, cDNA合成
(3) DNA polymerase with 3’-- 5’exonuclease and themal-resistance
dATP dGTP dCTP dTTP
Mg2+
buffer
3.Reaction . denature annealing extension
dsDNA ssDAN 37-72 °C 72 °C 92-96°C
50-58 °C
PCR reaction mixture
1.缓冲液 buffer (1×)
50Mm (大于500bp) (70-100Mm , 小于500bp) KCl 引物退火 annealing 10mM Tris·HCl pH 8.3-9.0 1.5mM MgCl2 (0.5-2.5) Stored in 20℃
2. dNTP
Final concentration 0.2mM (即饱和浓度) pH7.0
0.9kb
1.1kb
2.9
4.8
6.3
Taq Pwo Long sys
酶 pfu T4 T7 Vent Taq Taq klenow
ห้องสมุดไป่ตู้
dNTP (mM) 0.1 2.15 3.5 0.5-1. 5 0.5-1. 5 16.6 1.5
pH 8.4 8.0 8.0 8.5 8.0 8.8 7.9
Mg + 2 1.5 5 2.5 7.5 5 10 10
4. Template
µg or ng 102 ----105 3×105 targets 3×105 3×105 9 ×106 106 1µg 人类单拷贝基因组DNA 10ng yeast DNA 1ng E.coli DNA 1ng 1kb DNA 1% M13 plaque
5. DNA polymerase
PCR reaction program
1. Frequently program 94-96℃ 预加热 10’’-几分钟/加酶 94℃ 30” 55℃ 30” 72℃ 1’ 72℃ 3-7min 4℃
2.Annealing temprature(退火温度) . (退火温度)
Tm-5℃ 特异性高,需高温 If high Tm (55℃-72℃),using two-step method 37℃ for rondom primers
Primer design
① ≥16bp, frequently 20-30bp ② Tm=81.5+16.61og[J+]+0.41(G+C)%-(675/N) N: length of strand J: concentration of monovalent ion If N=20 G+C%=55% [J+]=60mmol/L then Tm=81.5-20.3+22.6-33.75=50.05
● ● ● ● ●
dNTP or enzyme的稳定性 末端产物抑制(pyrophosphate焦磷酸, dsDNA) 非特异性竞争(非特异性产物or primer-dimer) 特异性产自身复性(>10-8M) 高浓度产物下, 变性不彻底.
5.Preparation of reaction solution .
5℃ 5min
Application of PCR in gene cloning
Adding RE sites 1.Primer designing (引入酶切位点) 切点的类型
A 5‘:8 base序列,其中3-4为附加,4-6为酶切点,增加 引物 长度,但酶切效果仍然不好。 B 若内部序列为已知, 则根据需要,可进行5‘端突变设计, , 5‘ 效果更好
(1) 常规 最后加酶,及矿物油 (2) 具3’-5’活性酶 A: template, primer, dNTP H2O B: buf, enzyme, H2O (3) 热起始 hot
start
下层:dNTP buf primer 中间:蜡珠(Ampli Wax PCR Qam100) 上层: template (Taq) ·先加下层液,再一粒蜡株,70℃ 10min ·迅速加30µl上层混合物
切点的位置 参见限制性内切酶部分
如EcoRI, 5’端有一个C, 在 2hr内可切割>90% HindIII 5’端有3个C, 在 2hr内可切割>10% 20hr内可切割>75%
Vector-T
Taq酶特性:在平端双链DNA的3‘端添加1个A
T载体:5‘端有1个T
效果好
Blunt-end ligation
(3) 反向 反向PCR (inverse PCR)
“正向”
“反向”
(4) 长片段的 长片段的PCR克隆 克隆
pH值变化,缓冲能力变化,以及锰离 子的促裂解,模板和产物DNA被损坏; 过长的分子变性困难;聚合酶与模板结 合难;错误率增加,错误的碱基反过来 导致聚合酶的失灵
多用混合酶(如Pwo, Pfu) 来扩增
A. Vent DNA poly (New England Blabs) Thermococcus litoralis, 85kDa Half-life: 1.8hr at 100℃ (使用MgSO4) but Taq 5min
B. Tli DNA poly (Promega)
85kDa 来源同Vent
用Klenow大片段或T4 DNA聚合酶消去3‘端的突出 碱基,获得平端PCR产物
Development of PCR
(1) Nest PCR ) 1st 2nd
(2) A/T cloning )
PCR产物:Taq poly 产物的3’末端 一般要在3’末端加一个核苷酸,通常为A 载体 pGEM-T, pGEM-T Easy (promega) pCR-3, pCR-II, pCR-3-Uni (半磷酸化)(Invitrogen) 核心 线性DNA
1-2U 早期使用klenow 94kDa
(1) Taq DNA polymerase, 75℃,
Thermus aquticus 嗜热水生菌,水生栖热菌 5’ -- 3’polymeration and 5’ --- 3’ exonuclease Taq half-time >2hr 92.5℃ 40min 95℃ 5min 97.5℃
C.Pwo
Pyrococcus woesei (BM--- Roche) 90kDa Half-life >2hr at 100℃ <5min for Taq
D. Pfu
Pyrococcus furiosus 激烈热球菌(stratagene)
E. 商用混合酶
pfu
• Ideal for high-fidelity amplification • Lowest error rate of any thermostable DNA polymerase studied • One of the most thermostable DNA polymerases known • Lack of extendase activity means no unwanted 3’ overhangs • Optimal for blunt-end PCR cloning • Optimum temperature near 75°C • 95% active after 1-hour incubation at 98°C • Native and recombinant versions available