MDD技术文档指导文件
MDD入门讲义

欧盟医疗器械法规要求——IEC 60601-1 解读和介绍主要内容- 欧盟法规介绍 欧盟法规介绍 - 协调标准 协调标准 - IEC 60601家族标准介绍 家族标准介绍 - IEC 60601-1 3.0/3.1版本主要的变化 版本主要的变化 - 全球主要国家标准执行和适用情况 全球主要国家标准执行和适用情况 - Q&A2法规和指令欧盟的主要法律文书(1) 法规(Regulation): - 是一种具有普遍适用性和总约束力的法令。
- 它们适用于所有成员国,包括成员国的自然人。
- 法规一经生效 法规一经生效, 一经生效,各成员国都必须执行, 各成员国都必须执行,没有必要再制定相应的本国法规。
没有必要再制定相应的本国法规。
- 它们可取代或优先于与之冲突的国内法规。
指令(Directive): - 需在成员国制定的转换期之后转变为国家法律的法令。
- 虽然对各成员国均有约束力 虽然对各成员国均有约束力, 对各成员国均有约束力,但对于实施指令的具体方式和方法, 但对于实施指令的具体方式和方法,各成员可以 各不相同, 各不相同,只要能达到指令所要求的目标。
只要能达到指令所要求的目标。
- 指令是针对成员国颁布的,不针对自然人3法规和指令指令(Directive)——解决方案 - 欧洲指令对于所覆盖区域的法规制定提供了法律层面的框架。
- 欧盟成员国必须将指令转化成本国法律,各国法律因此保持协调。
- 在指令覆盖区域的产品必须符合所有适用的欧洲指令的要求, 指令覆盖区域的产品必须符合所有适用的欧洲指令的要求,并附加CE标志 以表明已符合要求 表明已符合要求 - 通过统一的合格评估过程使得产品在全欧洲上市。
CE标志代表了符合性,是一个准入的门框,而不是一个宣称质量的标识或通 标志代表了符合性 过测试的标识。
然而,法规要求仍然存在一些细小的差异,例如:各国对语言的要求。
4医疗器械适用指令医疗器械指令 (MDD) 93/42/EEC (过渡期截至1998年6月) 有源植入医疗器械指令( 源植入医疗器械指令(AIMD) 90/385/EEC (过渡期截至1995年1月) 体外诊断医疗器械指令( 外诊断医疗器械指令(IVDD) 98/79/EC (过渡期截至2003年12月)5MDD指令有三个主要方面需要特别关注: •安全性 (使用者、患者和公众) •有效性 (发挥预期的临床作用) •可重复性(制造过程)6定义和范围设备、 软件、材料或 “医疗器械”是指可单独或组合使用的所有工具、仪器、设备 设备 、软件 其他物品,包括制造商生产的专门用于诊断和/或治疗所必需的软件,制造商意 意 图将其用于人体,以: 图将其用于人体 ——诊断,预防,监测,治疗或减轻疾病, ——诊断,监测,治疗,减轻伤痛或残疾或予以补偿, ——调查,更换或改变解剖或生理过程, ——节育, 以及没有在人体内/上实现其主要效用的药物、免疫或代谢方式,但可通过这种 方式来协助实现其功能。
分子鉴别诊断(MDD)技术在呼吸道病原体检测中的应用

分子 鉴别诊 断 【 MDD) 术在 呼 吸道病原 体检 测 中的应用 技
韩卫 宁 ,张正姬 雅 雪蓉 , ,季 伟 衡 , 伟 夏 , 瑜 许 元根 , ,史智扬
(苏州市疾病预 防控制 中心检验科 , 苏州大学 附属 儿童医 院呼 吸科 , 。 苏州 大学 附属 第一 医院呼 吸内科 ,江苏 苏州 2 50 ; 104
H N e nn , H N hn - , AX ern J e, A W i ig Z A GZ eg i Y u— g, IW i — j o
HENG i We ,XI u,XU an g n,S AY Yu — e HIZhiy g -an
La rt r De a t e , S z o o aoy p rm nt u h u Ce t r o Die s s n e f r s a e Co r l nd nto a Pr v n in,S z o 5 0 e e to u h u 21 0 4, Chna i
江 苏省 疾 病 预 防 控制 中心 , 苏 南 京 20 0 ) 江 10 0
Th p lc to fm o e u a if r n i e a p i a i n o l c l r d fe e t— a d a n s s l to m i de tfc to l i g o e p a f r n i n i a i n i o he r s i a o y p t g n ft e p r t r a ho e s
v n e Te PCR ( re e r h d a cd m- Tag t n i e mut lx c l e PCR )wi i p t h
急性 呼吸 道 感 染是 全 世 界 范 围的 常见 病 、多 发
MEDDE V2.7.1(中文版翻译)

MEDDEV.2.7.12003年4月医疗器械指南临床数据评价:制造商和认证机构指南说明本指南为一系列与EC—医疗器械指令应用问题相关的指南中的一部分。
并不具有法律约束力。
该指南在经过与各个利益方(主管机构、欧盟委员会服务处、工业、其他有兴趣的团体)进行深入协商之后谨慎拟定而成,期间对中期草案进行了传阅,而且部分意见还为本文件所采纳。
因此,本文件反映出了来自医疗器械行业的利益团体代表所持的立场。
Commission européenne, B-1049 Bruxelles / Europese Commissie, B-1049 布鲁塞尔–比利时. 电话:(32-2)299.11.11. 传真:(32-2)296 70 13. 电子邮件:entr-medical-devices@cec.eu.int1. 引言与目的本文件之主要目的是为制造商提供审核和分析临床数据方面的指导意见,并且在当认证机构对制造商临床数据评价进行审核的时候,作为90/385/EEC(AIMD)[1]和93、42、EEC(MDD)[2]所规定的符合性评估程序的一部分提供给认证机构。
本文件还通过提供期望方面的指导意见给予制造商一定帮助。
2. 背景制造商必须按照指令中的规定,论证其预期目的和就其实现的安全性与性能所做出的声明。
根据一般规律,上述论证需要临床数据的支持(附录X,MDD 1.1)MDD附录X和AIMD附录7中所述的临床数据评价与以下规定之间存在密切关系:MDD 附录I:通用要求第1节和第3节;AIMD附录1:通用要求第1节和第2节。
还应注意附录I之I.6(MDD)和附录1之1.5(AIMD)。
3. 术语解释在本指南中:3.1 临床数据是指与器械临床安全和性能各方面有关的数据。
必须包括来自以下来源的数据:(i)与待评估器械市场经验有关的已发表和(或)未发表数据;或能够证明与上述待评估器械等价的类似器械;或(ii)相关器械的前瞻性临床研究;或(iii)临床研究结果,或针对能够证明与上述待评估器械等价的类似器械的科技文献报道的其他研究结果。
mdd和mdr的条款

MDD和MDR的条款一、引言随着医疗技术的快速发展和全球化进程的加速,医疗器械在医疗保健领域中的作用日益突出。
医疗器械的监管要求也随之变得严格和复杂。
欧洲联盟(EU)对医疗器械实施了医疗器械指令(MDD)和医疗器械法规(MDR),以确保医疗器械的安全性和有效性。
本文将深入探讨MDD和MDR的主要条款、特点和差异,以帮助医疗器械制造商、供应商和用户更好地理解和遵守相关法规。
二、MDD和MDR概述MDD是欧盟关于医疗器械的基本法规,于1993年正式实施,并进行了多次修订。
MDD的主要目的是确保医疗器械在上市时具有足够的性能和安全性,为患者提供可靠的治疗。
MDR是欧盟最新的医疗器械法规,于2017年正式实施,取代了MDD。
MDR旨在确保医疗器械在整个生命周期内都符合高标准的安全性和有效性要求,并加强了对医疗器械的监管。
三、MDD的主要条款MDD的主要条款包括以下几个方面:1.医疗器械的定义和分类:MDD明确了医疗器械的定义,并将医疗器械分为不同类别,以便对不同类型的医疗器械实施不同的监管要求。
2.符合性评估:MDD要求医疗器械必须通过符合性评估程序才能上市销售。
符合性评估机构必须是欧盟授权的公告机构之一。
3.CE认证:为了满足MDD的要求,医疗器械必须获得CE认证标志,证明其符合相关指令的要求。
CE认证标志是欧盟产品安全性的象征。
4.上市后监督:MDD要求制造商对已上市的医疗器械进行持续监督,以确保其安全性和有效性。
制造商必须建立有效的追溯系统,以便对医疗器械进行召回和追溯。
5.临床数据和性能评估:MDD要求制造商提供充分的临床数据和性能评估报告,以证明医疗器械的有效性和安全性。
四、MDR的主要特点与变化与MDD相比,MDR的主要特点与变化包括以下几个方面:1.范围扩大:MDR扩大了监管范围,涵盖了所有类型的医疗器械,包括体外诊断医疗器械、有源植入式医疗器械等。
2.全生命周期监管:MDR要求对医疗器械进行全生命周期监管,从研发、生产、上市到退役等各个环节都必须符合相关要求。
mdd对质量体系的要求_解释说明以及概述

mdd对质量体系的要求解释说明以及概述1. 引言1.1 概述本文旨在探讨面向模型驱动开发(Model-Driven Development,简称MDD)对质量体系的要求,并解释其背后的原因和意义。
质量体系是指为确保产品或服务达到预期质量水平而建立和维护的一系列组织、流程和方法。
MDD作为一种软件开发方法,强调基于模型的系统设计和实现,它对质量的要求在不同层面上有所不同。
1.2 文章结构本文主要分为五个部分。
引言部分是对整篇文章进行概述,介绍MDD对质量体系的要求及其影响和优势。
第二部分将介绍MDD的基本概念,并对质量体系进行解释说明。
接着,在第三部分中详细解释MDD对质量体系的具体要求,包括与质量管理原则的关联、组织结构和责任、过程控制与改进等方面。
第四部分将概述MDD对质量体系带来的影响和优势,包括提高产品质量和符合性能力、增强组织运作效率和灵活性以及实现持续改进与客户满意度提升等方面。
最后,第五部分对全文进行总结,并展望未来发展趋势及提出相应建议。
1.3 目的本文的目的是通过深入研究MDD对质量体系的要求,帮助读者了解MDD方法论与质量管理之间的关联,并认识到采用MDD开发方法所带来的质量改进机会。
同时,本文还将探讨MDD对组织和流程产生的影响,以及经济效益和用户满意度等方面的优势。
通过这些内容的介绍,读者可以更好地理解和应用MDD方法来提高软件开发过程中的质量水平。
2. MDD对质量体系的要求2.1 MDD简介在理解MDD(Model Driven Development,模型驱动开发)对质量体系的要求之前,首先需要了解MDD的基本概念。
MDD是一种软件开发方法论,它强调通过使用抽象模型来指导和支持软件开发过程中的各个阶段,从而提高开发效率和软件质量。
在MDD中,系统设计、实现和测试等活动依赖于构建并维护系统的模型。
2.2 质量体系概念解释质量体系是指以确保产品或服务符合特定标准和要求为目标的组织内部架构和流程。
公告机构技术文件(MDD)评估指南

NBOG’s Best Practice Guideapplicable for AIMDD, ⌧ MDD, and IVDD 2009-4 Guidance on Notified Body‘s Tasks of Technical Documentation Assessment on a Representative Basis1 IntroductionThe Directive 93/42/EEC concerning medical devices (MDD) contains possible conformity assessment procedures in order to CE mark devices. Up to now, there has been inconsistency in the way Notified Bodies (NB) have performed the assessment of technical documentations for Class lla and Class llb products following quality system conformity assessment routes.The Directive 2007/47/EC, covering the revision of the MDD, includes additional requirements for review of technical documentation for all Class IIa and Class IIb medical devices covered by quality system assessment routes.2 ScopeThis guideline has been prepared for NBs on how to assess the technical documentation on a representative basis according to the Directive 2007/47/EC. Attention was given to the sampling size and depth of the assessment. It will also be useful for Designating Authorities in monitoring the performance of their NBs in this area.3 Definitions3.1 Device subcategory (Directive 2007/47/EC, Art. 2, Paragraph 1 (iii), point (l) and Directive93/42 EEC Art 1 Paragraph 2 (l))A set of devices having common areas of intended use or common technology.3.2 Generic device group (Directive 2007/47/EC, Art. 2, Paragraph 1 (iii), point (m) andDirective 93/42 Art. 1 Paragraph 2 (m))A set of devices having the same or similar intended uses or commonality of technology allow-ing them to be classified in a generic manner not reflecting specific characteristics.4 Directive Requirements4.1 Class IIa devicesFor Class IIa devices the requirements are covered in Directive 2007/47/EC at Annex ll, Para-graph 2 (i), Paragraph 5 (f), and Paragraph 6 (e). The Directive 93/42/EEC is amended and An-nex II 7.2, Annex V 6.2 and Annex VI 6.2, states that “For devices in Class IIa the notified body shall assess ... the technical documentation … for at least one representative sample for each device subcategory for compliance with the provisions of this Directive.”4.2 Class IIb devicesFor Class IIb devices the requirements are covered in Directive 2007/47/EC at Annex ll, Para-graph 2 (i). The Directive 93/42/EEC is amended and Annex II 7.3 states that “For devices in Class IIb the notified body shall assess, as part of the assessment in Section 3.3, the technical documentation as described in Section 3.2 (c) for at least one representative sample for each generic device group for compliance with the provisions of this Directive.”4.3 Concept of the reviewsDirective 2007/47/EC, Recital (22) introduces the concept of the reviews of Class lla and llb de-vices: “… The depth and extent of this review should be commensurate with the classification of the device, the novelty of the intended treatment, the degree of intervention, the novelty of the technology or construction materials, and the complexity of the design and/or technology. This review can be achieved by taking a representative example of design documentation of one or more type(s) of devices from those being manufactured. Further review(s), and in particular the assessment of changes to the design that could affect conformity with the essential require-ments, should be part of the surveillance activities of the notified body.”Directive 93/42/EEC is amended and in Annex II 7.4, Annex V 6.3 and Annex VI 6.3, there are rationales provided for the sampling: “In choosing representative sample(s) the notified body shall take into account the novelty of the technology, similarities in design, technology, manu-facturing and sterilisation methods, the intended use and the results of any previous relevant assessments (e.g. with regard to physical, chemical or biological properties) that have been carried out in accordance with this Directive. The notified body shall document and keep avail-able to the competent authority its rationale for the sample(s) taken.”4.4 Requirements for surveillance assessmentsDirective 2007/47/EC Annex ll, Paragraph 2 (i), Paragraph 5 (f), Paragraph 6 (e) and Para-graph 6.4 introduces requirements for surveillance assessments. The Directive 93/42/EEC is amended and in Annex II 7.5, Annex V 6.4 and Annex VI 6.4 it is stated: “Further samples shall be assessed by the notified body as part of the surveillance assessment...”.5 TimingDirective 2007/47/EC Article 4 states Member States “… shall apply those measures from 21 March 2010”.Following the adoption of Directive 2007/47/EC, from 21 March 2010 at the latest, for Class IIb devices following the Annex II assessment route and Class IIa devices following the Annex II, V or VI assessment route there is a specific requirement for technical documentation to be reviewed by the NB on a sample basis. At any rate and at the latest, the first surveillance fol-lowing 21 March 2010 will need to address this element.After the 21 March 2010 new certificates can be issued only according to the new requirements. For existing certificates issued prior to this date please see “Interpretative document of the Commission's services Implementation of Directive 2007/47/EC amending Directives 90/385/EEC, 93/42/EEC and 98/8/EC” [1] for details. In this document the Commission’s ser-vices call upon the member states to require Notified Bodies to ensure that surveillance audits are performed at least once a year as specified in the standard EN ISO /IEC 17021 [2].6 Depth of AssessmentThe assessment of each set of technical documentation selected, as a minimum, should con-sist of a review of– the intended use including confirmation that the product is a medical device and it’s correct classification,– the validity of the essential requirements checklist, especially when harmonized standards have not been applied in full,– risk management file,– pre-clinical data (studies in animal models, biocompatibility, technical performance tests etc.),– clinical evaluation in accordance with 93/42/EEC Annex X (Note the NB review should be as per MEDDEV 2.7.1 [3]),– information supplied by the manufacturer (label, instructions for use),– declaration of conformity or the draft thereof,– other technical documentation based on risk,– and should be done according to the respective parts of NBOG BPG 2009-1 [4]. Consequently, there will be one part of the assessment performed within the normal course of the audit but in addition, file reviews will be necessary.The time spent on the review and the level of expertise used should be proportionate to the risk and complexity of the devices in question. For the more complex devices this may require a detailed technical review. If such an in depth assessment is not considered necessary then a justification should be available.Depending on the risk and complexity of the device(s), different experts may be involved. Therefore, NBs may not be able to perform the complete assessment on site.7 ReportingFor those elements covered the report produced for each technical documentation reviewed should be in line with the NBOG guidance on Design Dossier Report Content [4]. This could be either as part of the audit report or separately as a supplement.Nonconformities/Corrective Action Requests should be raised in the normal manner, including those raised against technical documentation under conformity assessment procedures according to Annex V or Annex VI.8 SamplingFor initial audits of devices in Class IIb the Directive states that the NB shall assess the tech-nical documentation for at least one representative sample for each generic device group (i.e. a set of devices having the same or similar intended uses or commonality of technology allowing them to be classified in a generic manner not reflecting specific characteristics). This is the same description as for “generic device groups” in EN ISO 15225 [5], which specifies the GMDN system.For initial audits of devices in Class IIa the Directive states that the NB shall assess the tech-nical documentation for at least one representative sample for each device subcategory (i.e. a set of devices having common areas of intended use or common technology). Since no similar description exists for subcategory/ies like for “generic device groups”, the grouped collectiveterms described in NBOG BPG 2009-3 [6], which define scope expressions for NBs, could be used for this.However, in practice it is expected that the NB identifies the complete range of Class IIa and IIb products manufactured and define a suitable sampling plan for initial audits. This should be done based on the numbers of generic device groups and grouped collective terms.It is considered that if large numbers of samples are required to be taken then the following plan should be used:For Class IIa devices: a sample of each group of scope expressions according to NBOG BPG 2009-3 [6].For Class IIb devices: a sample of generic device groups, referenced by different GMDN preferred terms, to the following plan:Up to 2 groups: a sample from each groupUp to 10 groups: a sample from 3 of these groupsUp to 20 groups: a sample from 5 of these groupsUp to 30 groups: from 7 groups a sampleN > 30 groups: from N/10 + 5 groups a sampleFor surveillance audits the NB should identify the complete range of Class IIa and IIb devices and group them according to preferred terms and grouped collective terms (scope expressions). For each certification period, and at least every 3 years, the NB should identify the technical documentations to be reviewed in line with the above sampling plan. As long as there is still a technical documentation not yet reviewed, at least one technical documentation should be sam-pled each year. This activity may or may not be done in conjunction with routine surveillance assessments. During subsequent certification periods appropriate plans should be drawn up to ensure that in the long run the majority, if not all, of the technical documentations will be assessed.9 Changes/New DevicesManufacturers introducing a new subcategory of Class IIa device or a new generic Class IIb device need to inform their NB who need to review relevant technical documentation prior to the products being placed on the market. For changes to existing designs the normal change requirements apply.Reference Directive 93/42/EEC Annex II, V and VIDirective 2007/47/ECSources [1][2] EN ISO/IEC 17021 : 2006 Conformity assessment – Requirementsfor bodies providing audit and certification of management systems[3][4][5] EN ISO 15225 : 2000/A1 : 2004 Nomenclature – Specification for anomenclature system for medical devices for the purpose of regula-tory data exchange[6]Devices Assessmentsdevice subcategory, generic device group, sam-assessment,Keywords conformitypling basis, technical documentationDate of issue July 2009。
MDD工作手册

每年9-10月
• •
草拟下一年MDD的AOP; 与各LU沟通渠道发展AOP,包括: – 当年业绩与次年目标 – 问题与机会点 – 策略和战术 – 人员和费用 MDD的AOP在与GM和其它功能部门沟通修正后,最终定稿,; MDD的各渠道拓展经理与MKT的各品牌经理共同进行YTD业务回顾,结合品牌经理提供 的市场促销方案和渠道拓展经理提供的渠道拓展方案制定MU季度行动计划(即QAP),由 GM/UM初审后发给FMDD和FMKT;
分渠道拓展的必要性
如何取得销量和利润的持续增长?
满足客户需求取得双赢
(各种不同渠道客户的集合)
C1公司: •不同渠道对销售系统有不 同的要求; •公司的资源需要整合 P1产品: •不同渠道有各自适合销售 的产品; C2客户: •不同渠道间客户需求不同; C3消费者: •消费者越来越个性化; C4竞争对手: •竞争越来越没有秘密可言;
步骤五: 项目执行/试点
优化销售资源配置
制定渠道拓展策略/战术和方案的 工作方法
Jan 29 5 12 19 26 2 Feb 9 16 23 1 8 Mar 15 22 29 5 Apr 12 19 26 3 May Jun July Aug 16 23 30 6 Sep 13 20 27 4 Oct 11 18 25 1 8 Nov 15 22 29 6 Dec 13 20 27 10 17 24 31 7 14 21 28 5 12 19 26 2 9
So as to cost-effectively reach and sustain maximum channel coverage and VPO. 从而以财务高效的方法达到并保持售点覆盖和单点销量的最大化!
目录
- 市场拓展部工作指导手册 - 市场拓展部的任务 - 市场拓展部组织架构
医疗器械 欧盟技术文档清单 MDD CE Checklist
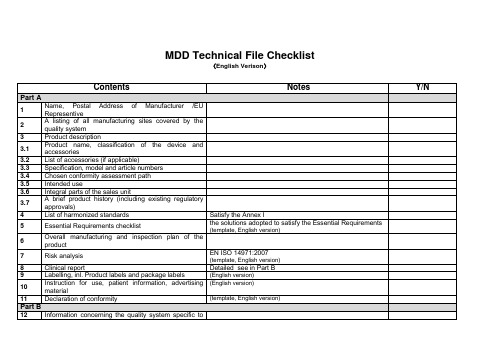
3.7
简要的产品历史(包括现有的管理审批)
4
适用的标准清单
符合医疗器械指令附录I
5
基本要求检查表
符合医疗器械指令附录I的方案
(有固定模板,需提交英文文件)
正在填写
6
产品的总体生产或质量控制方案
7
风险分析
EN ISO 14971:2007
有固定模板,需提交英文文件
正在索要
8
临床报告
详细的临床数据见Part B
9
Labelling, inl.Product labels and package labels
(English version)
10
Instruction for use, patient information,advertising material
(English version)
11
Declaration of conformity
MDDTechnical FileChecklist
(English Verison)
Contents
Notes
Y/N
Part A
1
Name, Postal Address of Manufacturer /EU Representive
2
A listing of all manufacturing sites covered by the quality system
13.2
Packaging and specification
(English version)
13.3
Description of the manufacturing processes
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
发布地点,日期:
签名:
7. 技术文档语言 一个成员国家可能要求技术文档的 A 部分使用官方语言,如果能懂可以不翻译。在需要翻译的情 况,允许文件拥有者额外的时间递交第一部分的内容给检查机构。
而且,关于翻译不能增加更多条件,例如要求有授权的翻译,官方翻译或类似要求。
注:本指令的信息仅作为生产商的指南,生产商仍需仔细阅读并理解 MDD.
2. 技术文档的来历
根据 MDD 93/42/EEC,所有的医疗器械必须符合基本要求附录 I 才能贴 CE 标志。技术文档则提供产 品符合基本要求的文件证据。
然而,技术文档这个词对制造商而言将比较费解,因为在 MDD 中未提到这个词。技术文档是技术 性文件的一个通称,用于证明产品符合基本要求。MDD 的不同附录将会略微不同地描述技术文 档。接下来便介绍每一个 MDD 附录如何描述技术文档。
附录 A) 全部的制造和检验计划 风险分析(见附录 B) 临床数据(见附录 C) 标签,如产品标签,说明书,病人信息,广告材料 合格声明
B 部分包含所有的检测和验证报告,产品有关的质量体系信息,产品的详尽描述如设计图,产品数 据参数,制造过程描述等。如果 B 部分是分散的,应建立一个控制目录,列出每个相关文件。
III 类医疗器械: -附录 II,包括第 4 部分(常指 II.4) -附录 III+附录 IV -附录 III+附录 V
IIb 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3) -附录 III+附录 IV -附录 III+附录 V -附录 III+附录 VI
IIa 类医疗器械: -附录 II,不包括第 4 部分(常指 II.3)
A 部分生产地必须在制造商处,在合格机构评审时在制造商处可以获得。如果制造商不在欧盟 范围内,A 部分必须在授权的欧盟代表处可以获得。B 部分可以独立于 A 部分,它的生产地接 近 A 部分,可以是制造商的另一个生产点,或位于海外的 OEM 供应商。
A 部分包含了大多数重要的与合格评定程序有关的技术数据的概括,包括: 制造商和欧盟代表名称和地址 质量管理体系覆盖的生产地点 产品描述,包括 产品标识(型号等) 预期用途的描述 产品附件(适用情况下) 销售单元的一体性部件(适用情况下) 产品及其附件的分类 选择的合格评审途径 产品的简要历史(包括已有的批准文件) 制造商采用的协调化标准清单和/或满足基本要求的解决方案(例如,基本要求清单-见
标准 欧洲标准委员会,CEN,CENELC 采用的标准用前缀 EN 进行识别。一旦一个标准在欧盟官方杂志发 表,就称为协调化标准。协调化标准清单将定期在欧盟官方杂志发表。
因为非欧洲的制造商对跟踪这些欧盟特殊标准存在困难,我们建议他们利用欧洲贸易组织,例如 IAPM,EUCLMED,或标准服务以获得这些信息。附录 D 包含标准组织,欧盟贸易组织,标准服务, 欧盟官方杂志来源的名称和地址,他们将帮助提供标准的现有状态和标准拷贝。
如果一个权威机构需要技术文档,技术文档的 A 部分必须立刻能获得。考虑到 B 部分的容量和格 式,可以稍微缓一点时间。技术文档 A 部分和 B 部分的详细内容将在本文第 6 部分进行说明。
6. 技术文档的内容和形式
概要
简而言之,技术文档所要求的信息包括:
(i)
证明产品符合基本要求的必要的技术信息
(ii)
8. 技术文档控制
一旦编制结束,技术文档应受控。虽然不必在文件受控中,但仍需某些程度的控制。文件必须随时 包含产品和支持数据的正确描述。因此,它必须联系于制造商工程变化系统,就如 510(K)和 PMA 的文件。如果产品发生变化,技术文档需要更新或评估。
技术文档控制失败将导致递交给合格机构的文件过期或数据矛盾。这将导致制造商因技术文档不充 分而错误使用 CE 标志而面临惩罚,。
定的稳定,不能危害医疗环境、危害患者、使 造商应考虑不良反应是否可接受。产品的寿命
5. 技术文档和权威机构
技术文档必须受权威机构支配以便于其检查 。这个要求来自欧盟 1985 年 5 月 8 日采纳的“技术协 调性和标准的新方案” ,并促进了新指令的产生。
而且,MDD 的附录中包含了如下要求:
-附录 II 的 4.1 提到“在产品生产后至少 5 年期限内,制造商必须确保 4.2 中所提到的文件受国家 权威机构的支配”
-附录 VII+附录 IV -附录 VII+附录 V -附录 VII+附录 VI
I 类医疗器械,含无菌和/或测量功能: -附录 VII+附录 IV -附录 VII+附录 V(无菌仪器必须采用这一途径) -附录 VII+附录 VI
I 类医疗器械,无无菌和/或测量功能: -附录 VII
合格评审途径 强调了为何附录 II,III,VII 涉及了技术文档(请见本文件 2.技术文档的来历)。名义 上,只有附录 II,III,VII 涉及了技术文档是因为所有的医疗器械必须使用附录 II,III 或 VII 以满足 11 章要求并使用 CE 标志。
基本要求: 毫无疑问,技术文档的灵魂是能证明符合基本要求的技术信息。根据 MDD 的第 5 章,符合基本要 求的前提是符合协调化标准。然而,实际上,并不是所有的医疗器械存在协调化标准。因此,在协 调化标准不存在时,使用其它存在的国际性标准,国家标准,一些贸易组织标准,例如 ISO, IEC, AFNOR, DIN, ANSI, AAMI, ASTM 等。制造商可以选择任何可接受的技术方法以符合基本要求。然 而必须注意,利用技术基本原理而非存在标准,将导致公告机构和合格机构的详细审查和时间上的 拖延。
品和包装的符合性。
施,包括安装报警装置。
IV. 评审标签和说明书(如果适用)。
-最后,告知用户因提供防护措施的弱点所带 来的残留危险。
V. 评审用于产品商业销售的最后放行的程 序
已销售器械 依靠产品的销售历史和上述 III 的内容。
3.
器械最后必须取得生产者期望获得的功能。器 这是性能要求。制造商必须证明器械符合特定
械设计,制造和包装应适合于第 1 条(2)(a)所规 要求。任何方案都反映这一点。
定的各项功能的发挥。
当制造商运行指令附录 II,V,VI 的质量体系时,
基本要求评审至少是质量体系的一部分
4.
在生产者确定的器械使用寿命期内,在正常使 制造商必须证明他已识别在确定的器械使用寿
用条件下,第 1,2,3 款指的各项性能应保持一 命期内,在正常使用条件下所产生的压力。制
附录 A 基本要求检查表 附录 B 风险分析 附录 C 临床数据
Байду номын сангаас
I.
总要求
1.
器械的生产和设计必须保证:按照其预定功能 1 和 2 条款要求器械的安全和有效。
和条件使用情况下,器械不会损害医疗环境、
对新器械和旧器械的方案不同。
患者安全、操作者或其它人员的安全和健康; 使用时的潜在危险与患者受益相比较可以为人 们所接受,并具有高水平的防护方法。
新器械
I. 评审设计概要,通常表现为产品参数的 设计方案,包括符合协调化标准的风险
2.
生产者的设计和制造方案,必须考虑在现有工
分析。
艺技术条件下遵守安全准则,生产者应:
II. 评审文献和自己拥有的类似经验
-首先,尽可能地降低甚至避免危险
III. 根据自己的标准和有关公布标准评价产
-其次,对无法避免的危险采取适当的防护措
医疗器械指令 93/42/EEC
技术文档指导文件
技术文档
1. 介绍
大部分新的指令需要制造商提供能证明产品符合指令基本要求的技术性文件。在指令中,技术性文 件通常指技术文档。尤其值得注意的是,在 MDD 中,每个医疗器械都应有技术文档。
虽然在 MDD 中对技术文档的内容有所描述,这里还将提供更为详尽的描述以帮助生产商理解和编 制技术文档。而且,符合本指令的技术文档将有助于公告机构和国家检测机构的评审。
因此,为简化起见,MDD 每个附录要求的用来证明满足基本要求技术性文件简称为技术文档。
3. 11 章及合格评审途径的回顾 完全理解技术文档需要快速浏览 MDD 的 11 章(合格评审途径)。11 章要求制造商满足 MDD 的某 个附录要求以获得 CE 标志。使用哪一个附录取决于仪器的分类。合格评审途径总结如下:
附录 III 附录 III 的第 2 部分提到“第 3 部分提到的文件需要评价抽样样品的符合性” 第 3 部分提到“需要 采用风险分析结果和标准清单(参考第 5 章)以满足基本要求”
附录 VII 附录 VII 的第 2 部分提到“制造商必须提供第 3 部分所说的技术文档”,第 3 部分进一步要求“需 要采用风险分析结果和标准清单(参考第 5 章)以满足基本要求”
附录 II 附录 II 的 4.1 部分提到“制造商必须向公告机构提交申请产品相关的设计文档的检查”
4.2 部分提到“申请必须包括 3.2(c)部分所说的文件,以评价产品是否符合指令要求,” 3.2(c)部分要求“产品的一般描述……包括适用的标准……以及为符合基本要求采取的方案的描 述。”设计文档是 III 类医疗器械的技术性文件,公告机构会全面审查。
-附录 III 的 7.3 提到“制造商和授权代表必须在产品生产后至少 5 年期限保留技术文档 ” -附录 VII 的 2 提到“制造商和授权代表必须编制文件,包括合格声明,在产品生产后至少 5 年期 限保留技术文档 ,便于国家权威机构检查”
产品在欧洲市场销售后,不管产品的最初产地,制造商或欧盟代表有义务保证自产品销售后可以获 得技术文档。
一个证明符合基本要求的简易方法是通过检查表,它系统列出了每个基本要求和证明符合性的方 法。这个检查表减轻了制造商,公告机构,合格机构的压力。因此,附录 A 包含了基本要求检查表 和另外的解释要求的指南。附录 A 的检查表可以扩展到更多栏目,包括要求的应用,证明符合性所 使用的标准,支持文件的位置,部分符合标准的技术原理,评论等。