能带计算
光子晶体能带计算

光子晶体能带计算
摘要:
1.光子晶体的概念
2.光子晶体原理
3.光子晶体能带计算方法
4.光子晶体在现代科技中的应用
5.总结
正文:
光子晶体是一种具有周期性结构的光学材料,其内部折射率不同,可以对光波进行散射和限制。
在光子晶体内部的波导可以具有非常尖锐的低损耗弯曲,这可以使集成密度增大多个数量级。
光子晶体原理是由苏联科学家
V.G.Veselago 在1968 年提出的左手介质理论,而美国物理学家D.R.Smith 在2000 年做出了人工左手介质。
光子晶体能带计算方法是通过研究光子晶体中的光子带隙,即在某一频率范围内的光波将发生反射,而不是通过晶体传播。
移除晶体结构中的部分砷化镓晶柱后,将产生适合带隙内频率的波导,随后光可以沿波导几何轮廓传播。
光子晶体在现代科技中的应用非常广泛,例如在集成光子学领域,光子晶体可以作为光波导、光开关、光调制器等光学器件。
此外,光子晶体还可以用于太阳能电池、LED 照明、光纤通信等领域。
总之,光子晶体是一种具有特殊光学性质的材料,其原理和能带计算方法为现代科技提供了新的解决方案。
能带理论(3)(紧束缚近似)

• 因J > 0,能带的最小值在 k 0,0,0
• 能带底的值为 • 能带的最大值在,
Emin s J 0 6J1
k 1, 1, 1
a
• 能带顶的值为
Emax s J0 6J1
• 能带宽度为 E Emax Emin 12J1
谢谢观看! 2020
i*(r Rm) 左乘,积分得到
ian i*(r Rm )U (r) V (r Rm )i (r Rm )dr
Ean
am i*(r Rm )U (r) V (r Rm )i (r Rm )dr m
引入变量
r Rm
(E i )an
考虑到U(r)为周期函数,即 上面方程中的积分式变为
m
s
在紧束缚态近似下,
E(k) i J0
J (Rs )eik.Rs
Rs 近邻
分裂的原子能级过渡成能带
• N个相同孤立 原子的分裂能 级,N重简并
• 原子靠近形成 晶体,简并能 级相互作用, 分裂形成能带
• 能带图上,不 同的N个k的 能级形成能带
comments
• 带宽取决于J,J积分取决于波函数交叠的多少 • 波函数交叠?波函数分布形状? • 内层电子分布区域大还是小?组成晶体后,能带宽
能带计算方法物理思想
• 各种能带计算方法基本上可分为
* 对晶体势场V(r)的不同近似 * 对组成晶体电子波函数的基函数的不同选取
• 根据不同的研究对象、根据计算条件作取舍 • 能带计算方法从构成晶体波函数的基函数上可
分成两大类:
* 紧束缚近似 * 近自由电子近似
• 两类近似的物理思想不同
近自由电子近似
把孤立原子的势场看成零级近似,而原子间相互作用看成微扰, 这种微扰是N重简并微扰,微扰后的状态是N个简并态的线性 组合。
超晶格能带计算 有效质量理论
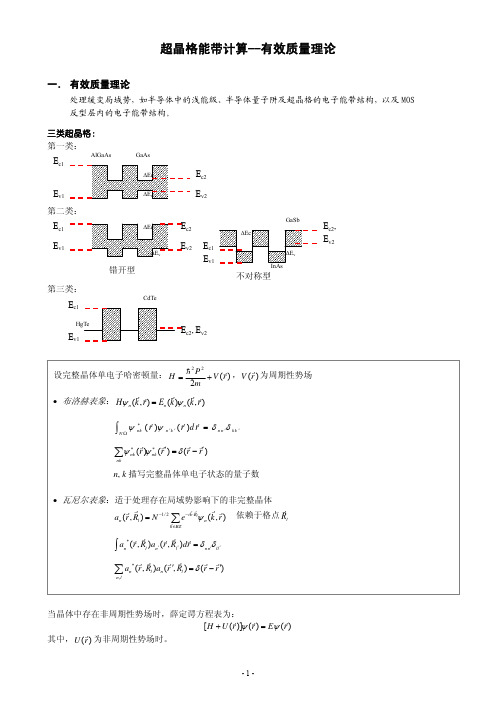
+
∂2 ∂y 2
+
∂2 ∂z 2
)
其中,m*是电子有效质量(导带底附近是各项同性的),假设了能量极值点在Γ点(
r k0
=
0 )。
有效质量方程为:
⎢⎡− ⎣
h2 2m∗
∂2 ( ∂x2
+
∂2 ∂y 2
+
∂2 ∂z 2
)
+
U
(rr)⎥⎤ ⎦
F
(rr
)
= [E
−
En (0)]F (rr)
波函数:
ψ (rr) = F (rr)unk0 (rr) = F (rr)usk0 (rr)
= 0 , ki2
=
2mi h2
(E −Vi )
其解的形式是沿 z 轴正负两个方向平面波的叠加:
-∞ ≤
z
<
z1: G0
=
A eik0 ( z−z1) 0
+
B e −ik0 ( z−z1) 0
z1 ≤ z < z2: ……
G = A e + B e ik1(z−z1)
−ik1( z−z1)
1
1
1
(注意取 z0 = z1)
注意此式中
Fj
(rr)
与
a
j
r (k
)
的关系[?]。
-4-
二. 导带的计算
1.导带不参与其他带的耦合
----单带模型的有效质量方程(抛物带模型)
导带底等能面是球面,抛物型能量色散关系为:
En
r (k )
=
En
(0)
+
h2k 2 2m∗
9.3 能带的计算

φ k = e ik ⋅ r u 0 ( r )
因为已考虑到离子实的作用势,上述函数比平面波函数更 因为已考虑到离子实的作用势, 接近于真正的波函数; 接近于真正的波函数;但这个近似解的能量对波矢的依赖 完全和平面波一样, 完全和平面波一样,因为 u0 (r ) 满足
1 2 [ p + U (r )]u0 (r ) = ε 0u0 (r ) 2m 这样 1 ℏ 2k 2 [ ( p 2 + ℏ 2 k 2 ) + U (r )]u0 (r ) = [ε 0 + ]u0 (r ) 2m 2m
15
< ε k >= −8.2 + 1.9 = −6.3eV
而自由原子价电子的能量为 −5.15eV; ; 相对于自由原子, 相对于自由原子,金属钠约有 1.1eV (实 实 验值 1.13eV) 的能量降低而具有稳定性
10
固体物理导论 固体物理导论 物理
第 9 章 费米面和金属
9.3 . 能带的计算
按第10章 此势应被屏蔽, 按第 章,此势应被屏蔽,每个 U(r) 的傅里叶分量应除以 电子气的介电常量 赝势比真实势弱得多, 赝势比真实势弱得多,但在外部区域中这两势的波函 数接近于全同
13
固体物理导论 固体物理导论 物理
第 9 章 费米面和金属
9.3 . 能带的计算
金属钠的赝势:空芯模型,并由托马斯金属钠的赝势:空芯模型,并由托马斯-费米介电函数屏蔽
φ k = e ik ⋅ r u 0 ( r )
具有布洛赫的形式, 的项时, 具有布洛赫的形式,但只有舍弃 k ⋅ p 的项时, 0 (r )才是上 u 述方程的解; 一般作为微扰处理[习题 习题8] 述方程的解; k ⋅ p 一般作为微扰处理 习题
能带的理论计算和WIEN2K软件的简介

WIEN2K程序的操作流程:前期准备
1、启动web 服务器:在运行WIEN2K 的计算机上启动用户界面web(在机 器上添加telnet,ssh),使用以下命令:w2web [-p xxxx]
2、w2web 服务器的连接:使用喜欢的WWW 浏览器连接w2web,定义正 确的端口数,比如netscape http://hostname where w2web runs:7890
平面波方法
正交化平面波方法
正交化平面波方法
赝势方法
优点:可选取最佳的赝势以简化能带计算。 缺点:不适合于价电子与芯电子难以严格区分的固体,例如稀土 金属和过渡金属。
缀加平面波方法
缀加平面波方法
缺点: (1)在建立Muffin-tin球的径向解时隐含了能量,它们的矩阵元都是能 量的函数,使久期方程成为能量的超越方程,需要另外用白洽计算来求 解能量本征值。 (2)当径向解的节点落在Muffin—tin球面上时,久期方程出现奇异 性。 (3)基函数的导数在球面上不连续。
线性缀加平面波方法
在Muffin-tin球内给APW基函数增加一对能量求导的项,使得球内径向函数 的解不再是能量本征值的函数,而代之以某个待选定的能量参数值,这 就是线性缀加平面波方法(LAPW)。
采用Muffin-tin势场的LAPW方法对密堆积结构的金属是很好的近似,但对 于非密堆积的开结构,如半导体、清洁的或有吸附物的表面、薄膜、空洞 材料、准低维材料等,用这样是势是不合适的。基于多极势的概念,提出 了一个新的方法,改进了对势的形状限制,对LAPW方法进行了修正和发展, 称之为全电势LAPW方法或FLAPW方法。
WIEN2K程序的操作流程:计算
计算的初始化: 通过“Execution initialize calc.”完成计算的初始化,从而可以给出初始化计算 所需的步骤
4平方米铜线能带多少瓦电器?如何计算?小方法告诉你!

4平方米铜线能带多少瓦电器?首先需要知道电线怎么计算。
计算电线的半径用求圆形面积的公式计算:电线平方数(平方毫米)=圆周率(3.14)×电线半径(毫米)的平方知道电线的平方,计算线直径也是这样,如:2.5方电线的线直径是:2.5÷ 3.14 = 0.8,再开方得出0.9毫米,因此2.5方线的线直径是:2×0.9毫米=1.8毫米。
计算电线的平方也用求圆形面积的公式来计算:电线的平方=圆周率(3.14)×线直径的平方/44mm²铜芯线长期且不发热的安全载流量为4x6=24A(16㎜²及以下铜芯线经验系数按1mm²×6A计算)。
单相电器功率:P=IUcosa,单相电器功率因素一般为0.75~0.8。
按最低功率因素计算。
由此可知P=24×220×0.75≈4KW三相电器功率P=√3UIcosa,三相电器功率因素一般为0.7~0.95。
同样按功率因素计算。
P=1.732×380×24×.07≈11KW如果导线过长,其导线的电阻及阻抗就不能忽视。
R=ρL/S(ρ:电阻率,铜芯线为0.0175。
L:导线长度,S:导线截面积)。
另外导线敷设是明敷还是暗敷及多线同管敷设的存在散热方面因素。
综合以上因素,4mm²用于单相时,允许承载电器功率应在4KW以下。
用于三相时,允许承载电器功率应在10KW“经验法”,我们用最熟悉的一种用电器具,那就是单相异步电动机。
我们经常做电工的都知道,一个平方毫米铜导线大约能带动1千瓦的电器,并保证导线在长期运行下不发热。
显而易见4平方毫米铜线能带大约4千瓦的电器并能保证长期稳定工作,这种方法简单明了不需要复杂的计算截流量、计算电流等,特别是对我们老百姓来说更是简化了使用方式,我在接线时经常按照这个经验去估算安全又可靠。
当然如果不处于长时间运行的情况下,我们也可以把负载放大一些,为了安全考虑最好不要超过6千瓦。
能带论计算方法简介

a
8
3、哈特利-福克近似
通过绝热近似,把电子的运动和原子核的运动分开,得到了多电子薛定谔方程:
引入哈特利波函数 : 通过哈特利-福克自洽场近似方法,将多电子的薛定谔方程简化为单电子有效势方程:
在哈特利-福克近似中,已包含了电子与电子的交换相互作用,但自旋反平行电子间的排 斥相互作用没有被考虑:在 r 处已占据了一个电子,那么在r’处的电子数密度就不再是 p(r’) 而 应该减去一点;或者说,再加上一点带正电的关联空穴,即还需考虑电子关联相互作用。
在弱周期场近似中,波函数由平面波叠加而成,要使波函数在离子实附近有振荡的特 点,平面波的展开式中要有较多的频率成分,因而收敛很慢,所以平面波方法计算固体能 带实际计算难以进行。
1940年 Herring 提出了OPW方法,取波函数为平面波和紧束缚波函数的线性组合, 并要求与离子实不同壳层紧束缚波函数正交,从而自然地兼顾了波函数在离子实附近以及 在离子之间应有的特征,求解时,往往只需要取几个正交平 面波,结果就很好了。
a
9
4、交换关联泛函的简化
在 Hohenberg-Kohn-Sham 方程的框架下,多电子系统基态特性问题能在形式上转化成有效单 电子问题。该计算方案只有在找出交换关联势能泛函的准确的、便于表达的形式才有意义。
在具体计算中常用 W.Kohn 和 L.J.Sham 提出的交换关联泛函局域密度近似是一个简单可行而又 富有实效的近似。其基本思想是在局域密度近似中,可利用均匀电子气密度函数来得到非均匀电 子气的交换关联泛函。
方法上的简化使大分子系统的研究成可能,酶反应机制的理论计算就是其中典型的实例, 如今,密度泛函方法已经成为量子化学中应用最广泛的计算方法,因此沃尔特·科恩获得了 1998年诺贝奖。
能带理论(2)(微扰计算、能带、带隙)

能带理论(2)(微扰计算、能带、带隙).ppt
能量修正
(二级)
波函数修正
(一级)
平面波 被周期势场散射
动能
例题:设晶格常数为a的一维晶格的周期性势场为
用自由电子近似的微扰论,近似地求出布里渊区边界/a处 的能隙 解:
把U(x)展开为复数傅立叶级数
我们发现傅立叶系数只有两个,即
而布里渊区边界/a正好是第一布里渊的边界,能级在此发 生分裂,分裂值为
考虑一个二维正方格子,其晶格势场为
ห้องสมุดไป่ตู้
考虑一个二维正方格子,其晶格势场为 用自由电子近似的微扰论,近似地求出布里渊区顶角( /a, /a )处的能隙
在U(x)展开为复数傅立叶级数时只有4个系数,即
V(1, 1), V(1, -1), V(-1, 1) 和V(-1, -1)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
四个输入文件
INCAR,KPOINT,POSCAR相似 注意:POTCAR
运行软件,得到优化的晶格常数:
自洽的电荷密度: 修改INCAR文件, 修改INCAR文件, 运行软件, 得到CHG, 得到CHG,CHG CAR文件
做非自洽计算:
• 修改INCAR文件: 将 参数ICHARG设为 11 • 修改KPOINTS输入 文件 • 运行VASP程序,从 输出文件EIGENVAL 中提出电子结构
首先我们要会想一下Si 首先我们要会想一下Si的 Si的 结构
பைடு நூலகம்
生成4个输入文件
Diamond Si 5.5 0.0 0.5 0.5 0.5 0.0 0.5 0.5 0.5 0.0 2 Direct 0.0 0.0 0.0 0.25 0.25 0.25
晶格常数 基矢a1 基矢a2 基矢a3 原胞中的原子个数 坐标系选为基矢构成的坐标系 基矢坐标系下原子的位置
第一原理电子结构计算程序:VASP
主要输入文件: 1、INCAR 2、POTCAR 3、POSCAR 4、KPOINTS
一、Si能带图 Si能带图 的计算
(1). 生成4个输入文件: POSCAR POTCAR INCAR KPOINTS (2). 优化晶格参数,求出能量最低所对应的晶格参数 (3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量 (4). 修改KPOINTS和INCAR输入文件,固定电荷密度,做非自洽 计算,得到输出文件EIGENVAL (5). 提取数据,画图
INCAR:此文件控制vasp进行何种性质的计算,以及 INCAR: 设置了计算方法中一些重要的参数。 POTCAR:赝势文件,可直接从赝势库中导出,有很多 POTCAR: 的种类,现在主要用paw势,即投影缀加波德赝势。 POSCAR:描述所计算体系的晶胞参数,原子个数及晶 POSCAR: 胞中原子的位置,以及分子动力学计算时原子的初 速度。 KPOINTS:设置布里渊区k点网格取样大小或能带结构 KPOINTS: 计算时沿高对称方向的k点
李晨辉 凝聚态物理
2011-10-28
1
密度泛函理论
密度泛函理论(Density Functional Theory,DFT),是基于 量子力学和玻恩-奥本海默绝热近似的从头算方法中的一类 解法,与量子化学中基于分子轨道理论发展而来的众多通 过构造多电子体系波函数的方法(如Hartree-Fock类方法) 不同,这一方法构建在一个定理的基础上:体系的基态唯 一的决定于电子密度的分布(Hohenberg-Kohn定理),从而 使得我们可以采用最优化理论,通过KS-SCF自洽迭代求 解单电子多体薛定谔方程来获得电子密度分布,这一操作 减少了自由变量的数量,减小了体系物理量振荡程度,并 提高了收敛速度,并易于通过应用HF定理等手段,与分子 动力学模拟方法结合,构成从头算的分子动力学方法。
优化晶格常数
自洽的电荷密度
general: System=fcc Si ENCUT=240 ISMEAR=-5 LORBIT=11
做非自洽计算, 求电子结构
•修改INCAR文件: 将参数ICHARG设为 11 • 修改KPOINTS输入文件 • 运行VASP程序,从输出文件OUTCAR中提出电子结构
INCAR输入文件: 程序控制参数
System =diamond Si ISTART = 0 ENCUT = 150.0 NELM= 200 EDIFF = 1E-04 EDIFFG = -0.02 GGA=91 NPAR=4 NSW=100 IBRION = 2 ISIF=2 ISYM = 1 LWAVE = F LCHARG = F
KPOINTS输入文件: 控制K-点的选取方式
K-Points 0 Monkhorst Pack 11 11 11 0 0 0
POTCAR输入文件: 赝势文件
US Si 4.00000000000000000 parameters from PSCTR are: VRHFIN =Si: s2p2 LEXCH = CA EATOM = 115.7612 eV, 8.5082 Ry GGA = -1.4125 -1.4408 .0293 -.9884 eV TITEL = US Si LULTRA = T use ultrasoft PP ? IUNSCR = 1 unscreen: 0-lin 1-nonlin 2-no RPACOR = 1.580 partial core radius POMASS = 28.085; ZVAL = 4.000 mass and valenz RCORE = 2.480 outmost cutoff radius RWIGS = 2.480; RWIGS = 1.312 wigner-seitz radius (au A) ENMAX = 150.544; ENMIN = 112.908 eV EAUG = 241.945 …………
二、GaAs的能带图 GaAs的能带图
(1). 生成4个输入文件: POSCAR POTCAR INCAR KPOINTS (2). 优化晶格参数,求出能量最低所对应的晶格参数 (3). 固定晶格参数, 求出能态密度(DOSCAR), 确定费米能量 (4). 修改KPOINTS和INCAR输入文件,固定电荷密度,做非自洽 计算,得到输出文件EIGENVAL (5). 提取数据,画图
GaAs的能带结构图
The End