用vasp计算硅的能带结构
vasp_能带结构和态密度图

能带结构和态密度图的绘制及初步分析前几天在QQ的群中和大家聊天的时候,发现大家对能带结构和态密度比较感兴趣,我做计算已经有一年半了,有一些经验,这里写出来供大家参考参考,希望能够对初学者有所帮助,另外写的这些内容也不可能全都正确,只希望通过表达出来和大家进行交流,共同提高。
MS这个软件的功能确实是比较强,但是也有一些地方不尽如人意的地方。
(也可能是我对一些结果不会分析所致,有些暂时不能解决的问题在最后一部分提出,希望大家来研究研究,看看有没有实现的可能性)。
能带结构、态密度和布居分析是很重要的内容,在分析能带结构和态密度的时候,往往是先作图,然后分析。
软件本身提供的作图功能并不是很强,比如说能带结构(只能带只能做point图和line图),不美观不说,对于每一个能带的走势也不好观察,感觉无从下手。
所以我一般用origin作图(右图是用origin做的能带图)。
能带结构和态密度的作图过程请参考我给大家提供的动画。
接下来我们先开看看能带结构的分析和制作!第一部分:能带结构这个部分打算先简单的介绍一下能带的基础知识,希望能对大家有所帮助,如果对能带了解比较深入的朋友,可以跳过这个部分内容,之中不当之处请勿见笑。
^_^第一个问题是:1、能带是怎样形成——轨道和一维体系的能带。
这是最基本的一个问题,我们要对能带结构进行分析,首先要知道它是如何来的。
其实能带是一种近似的结果(可以看成一种近似),是周期边界条件(bloch函数)下的一种近似。
先来看看一个最简单的问题,非周期体系有没有能带结构?答案是没有的,大家可以试试:①建一个周期的晶胞②选择build菜单下的symmetry子菜单下的none periodic superstructure去掉周期边界条件性③看看还能够运行吗?运行(run)按钮变灰了,不能提交作业了。
这说明什么问题?这说明这个CASTEP这个模块不能计算非周期的体系,另外可以参考MS中的DMOL模块,它可以计算非周期系统,虽然可以计算周期系统,但是仍不能计算能带,大家可以试试,看看property中的band structure能不能选上,一定不能!!^_^从这里,我们可以得到一个结论,对于单个原子(分子、单胞)如果不加上周期边界条件,是无法获得能带结构的。
VASP计算实例

VASP计算实例目录一、氢气分子H2键长的计算 (3)1.基本文件 (3)2.赝势类型的选择 (3)3.截断能ENCUT参数的选择 (4)4.KPOINTS参数选择 (5)5.对晶格常数进行优化 (6)二、Si晶体晶格常数计算 (8)1.赝势类型选择 (8)2.截断能(ENCUT)参数的选定 (9)3.KPOINTS参数的选定 (11)4.SIGMA参数的选定 (12)5.晶格常数计算结果 (13)三、Si元素单原子能量计算 (14)1.由内聚能倒推单原子能量 (14)2.基本文件 (15)3.单原子能量计算 (15)四、Si的VASP力学常数计算 (16)1.计算所需文件 (16)2.计算与数据处理 (17)3.计算所用到的公式: (18)五、SI晶体的电子结构 (19)1.采用VASP计算能带的步骤 (19)2.电荷分布计算结果 (20)能带计算和结果 (21)3.态密度计算和结果 (21)六、Si晶体介电函数和光学性质的计算 (22)1.计算步骤 (22)2.用到的文件 (23)3.计算结果 (26)七、VASP的声子谱计算 (29)1.计算步骤 (29)2.基本文件 (30)3.声子谱、声子态密度计算和结果 (33)4.热学性质计算和结果 (34)八、化合物co2键长计算 (35)1.计算步骤 (35)2.基本文件 (35)一、氢气分子H2键长的计算1.基本文件准备基本文件INCAR、POTCAR、POSCAR、KPOINT以及脚本文件encut、k、optimize2.赝势类型的选择输入文件如下其中参数要靠经验初选INCAR:System = F2ISTART = 0ICHARG = 2NELMDL = 5ISMEAR = 0SIGMA = 0.1PREC = AccurateKPOINTS:Automatic meshM1 1 10 0 0POSCAR:O115.0 0.00 0.000.00 14.0 0.000.00 0.00 13.01D0.00 0.00 0.00分别选用五个贋势文件进行计算。
vasp学习
vasp学习(内部资料)§1, Si 的优化和能带结构、态密度计算练习。
一, 基本流程。
1,建立一个Si的目录。
mkdir Si2, 准备四个输入文件:INCAR、POSCAR、KPOINTS、POTCAR。
INCAR: vasp输入控制文件。
POSCAR: 坐标文件。
包括原胞的基矢。
KPOINTS:k点选取。
POTCAR:原子的赝势。
1)INCAR文件的准备INCAR 输入文件:ISTART = 0ENCUT=400 !该参数需要测试ICHARG = 2ISMEAR = -5EDIFF = 1.0E-06#EDIFFG = -0.001NSW = 0IBRION = -1ISIF = 22), POSCAR 文件的准备。
这个可以从Material Stutio 建模得到。
点击File, 选择import Document, 在structure里找到Semiconductor,选择Si.msi。
即找到Si的结构。
输出原子坐标,在MS中点击Build-->symmetry-->primitive cell (对于已经是原胞的情况,这一步省略)在点击Build-->symmety->make P1.再点击file-->export,选择输出文件为Si.cif。
(必须选择cif格式)输出以后,用文本文档的方式打开Si.cif。
在Si.cif中看到这样几行,Si1 Si 0.00000 0.00000 0.00000 0.00000 Uiso 1.00Si2 Si 0.25000 0.25000 0.25000 0.00000 Uiso 1.00这就是Si的原胞中原子的坐标。
根据POSCAR需要进行编辑。
(在vi中可以用列模式进行编辑。
“Ctr+v”进入列模式,键盘上下左右箭头进行区域选择,“ctr+p”粘贴选中的内容,“d”删除选中的内容。
)原胞的基矢可以通过在MS中点击原胞的白色边框,在MS 的左侧的Properties 一栏中,将出现原胞的详细信息。
VASP计算DOS和能带

VASP计算DOS和能带个人总结一:VASP计算DOS和能带1.计算DOS①POSCAR②POTCAR③KPOINTS(建议以Gamma为中心取点,通常K×a≈45即可)④INCAR(越简洁越好)第一步:结构优化SYSTEM=**ISTART=0ENCUT=500(最好对其进行测试)EDIFF=1E-5EDIFFG=-0.01NSW=100ISIF=2IBRION=2【优化后计算DOS可以一步完成,也可以分为两步来完成,主要是计算量涉及到计算时间的差别】第二步:静态自洽(此时可稍微降低K点数,用第一步优化得到的CONTCAR作为POSCAR进行计算)SYSTEM=**ISTART=0PREC=AccurateEDIFF=1E-5EDIFFG=-0.01ENCUT=500ISMEAR=-5LCHARG=.TRUE.注意:此时得到的E-feimi是准确的,需要记下(grep ‘E-fermi’OUTCAR)第三步:非自洽计算(采用高密度K点)SYSTEM=**ISTART=1ICHARG=11LMAXMIX=2/4/6(VASP官网原话:If ICHARG is set to 11 or 12, it is strongly recommened to set LMAXMIX to twice the maximum l-quantum number in the pseudpotentials. Thus for s and p elements LMAXMIX should be set to 2, for d elements LMAXMIX should be set to 4, and for f elements LMAXMIX should be set to 6)PREC=AccurateEDIFF=1E-5EDIFFG=-0.01ENCUT=500(截断能最好与上一步保持一致)ISMEAR=-5LORBIT=10/11(推荐11,可以得到能级分裂的数据)优化后计算DOS一步完成:(采用高密度K点)SYSTEM=**ISTART=1PREC=AccurateEDIFF=1E-5EDIFFG=-0.01ENCUT=500ISMEAR=-5LORBIT=10/112.计算能带①POSCAR②POTCAR③KPOINTS:使用Line-mode格式,给出高对称性K点之间的分割点数,分割越密,路径积分就越准确。
第一原理电子结构计算程序:VASP

0.866 0.0
B A B A
(2). 优化晶格参数,求出能量最低所对应的晶格参数
wurtzite晶体含有两个内部自由度, 晶格参数优化过程要比立方 结构费时
CoO: a=2.98, c/a=1.735, u=0.367
8
8
4
4
E (eV)
0
0
-4
-4
-8
-8
-24
A
L
M
Γ
A
H
K
Γ
-24
A
L
M
第一原理电子结构计算程序:VASP
• 程序原理 • 输入文件 • 输出文件 • 应用
输入文件
POTCAR KPOINTS POSCAR INCAR
pseudopotentail file Brillouin zone sampling structural data steering parameters
r 1r a1 = a( i − 2 r 1r a2 = a ( i + 2 r r a3 = ck
3r j) 2 3r j) 2
r 2π r (i − b1 = a r 2π r (i + b2 = a r 2π r b3 = k c
3 3 3 3
r j) r j)
r r r Γ = 0b1 + 0b2 + 0b3 = (0,0,0) 1 r r 1 1 K = (b1 + b2 ) = ( , ,0) 3 r 3 3 M = 0.5b1 = (0.5,0,0) r A = 0.5b3 = (0,0,0.5) r r r H = 0.5b1 + 0.5b2 + 0.5b3 = (0.5,0.5,0.5) r r r L = 0.5b1 + 0b2 + 0.5b3 = (0.5,0,0.5)
VASP计算能带

VASP计算能带量子化学网版权所有/Experience/CommonSoftwares/VASP/Electroni cCalc/200602/1043.htmlVASP Version : 4.6在此文中,我将用硅晶体作为实例,来说明如何用VASP4.6来计算固体的能带结构。
首先我们要了解晶体硅的结构,它是两个嵌套在一起的FCC布拉菲晶格,相对的位置为(a/4,a/4,a/4), 其中a=5.4A是大的正方晶格的晶格常数。
在计算中,我们采用FCC的原胞,每个原胞里有两个硅原子。
VASP计算需要以下的四个文件:INCAR(控制参数), KPOINTS(倒空间撒点), POSCAR (原子坐标), POTCAR(赝势文件)为了计算能带结构,我们首先要进行一次自洽计算,得到体系正确的基态电子密度。
然后固定此电荷分布,对于选定的特殊的K点进一步进行非自洽的能带计算。
有了需要的K点的能量本征值,也就得到了我们所需要的能带。
步骤一.—自洽计算产生正确的基态电子密度:以下是用到的各个文件样本:INCAR 文件:SYSTEM = SiStartparameter for this run:NWRITE = 2; LPETIM=F write-flag & timerPREC = medium medium, high lowISTART = 0 job : 0-new 1-cont 2-samecutICHARG = 2 charge: 1-file 2-atom 10-constISPIN = 1 spin polarized calculation?Electronic Relaxation 1NELM = 90; NELMIN= 8; NELMDL= 10 # of ELM stepsEDIFF = 0.1E-03 stopping-criterion for ELMLREAL = .FALSE. real-space projectionIonic relaxationEDIFFG = 0.1E-02 stopping-criterion for IOMNSW = 0 number of steps for IOMIBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CGISIF = 2 stress and relaxationPOTIM = 0.10 time-step for ionic-motionTEIN = 0.0 initial temperatureTEBEG = 0.0; TEEND = 0.0 temperature during runDOS related values:ISMEAR = 0 ; SIGMA = 0.10 broadening in eV -4-tet -1-fermi 0-gaus Electronic relaxation 2 (details)Write flagsLWAVE = T write WAVECARLCHARG = T write CHGCARVASP给INCAR文件中的很多参数都设置了默认值,所以如果你对参数不熟悉,可以直接用默认的参数值。
用VASP计算硅的能带 (改进稿)
输入文件EIGENVAL和syml到程序pbnf.x,可得到输出文件bnd.dat和highk。将bnd.dat和highk中的数据导入到Origin,即可画出如图2所示的硅的电子能带结构图。
-0.02022028
0.8
-0.05656195
作出EENTRO和SIGMA的关系图,如图8所示。由图可见,在SIGMA=0.2时,EENTRO就已经非常接近于0了,故SIGMA的值可优化为0.2。
图8:EENTRO和SIGMA的关系图
POTIM = 0.25
NSW = 100
EDIFFG = -1E-2
ISMEAR = 0
SIGMA = 0.1
PREC = Accurate
ISIF = 2
Si-fcc
5.1
0.0 0.50.5
0.5 0.0 0.5
0.50.50.0
2
Direct
0.00.00.0
0.250.250.25
#采用PAW_PBE赝势
(一)优化晶格常数
只需要优化一个晶格常数a。
首先准备四个文件:INCAR,POSCAR,POTCAR,KPOINTS,内容分别如下
INCAR
POSCAR
POTCAR
KPOINTS
system = Si-fcc
ISTART = 0
ICHARGE = 2
ENCUT = 300
EDIFF = 1E-5
IBRION = 2
用VASP计算晶体硅的电子能带结构
用VASP计算硅的能带

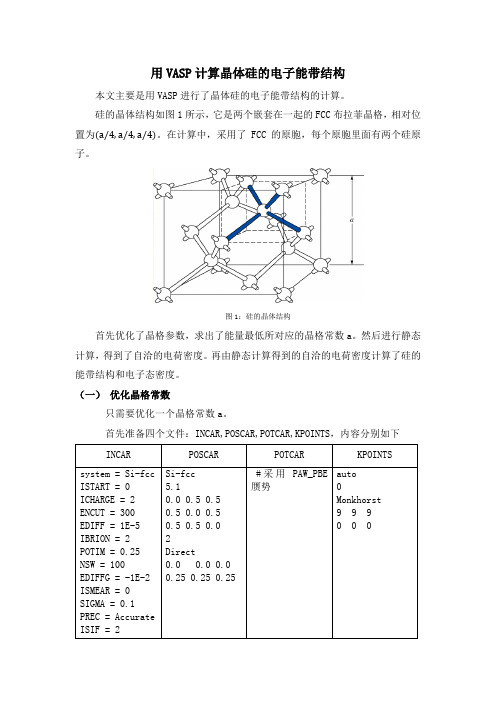
用VASP计算晶体硅的电子能带结构本文主要是用VASP进行了晶体硅的电子能带结构的计算。
硅的晶体结构如图1所示,它是两个嵌套在一起的FCC布拉菲晶格,相对位置为。
在计算中,采用了FCC的原胞,每个原胞里面有两个硅原子。
图1:硅的晶体结构首先优化了晶格参数,求出了能量最低所对应的晶格常数a。
然后进行静态计算,得到了自洽的电荷密度。
再由静态计算得到的自洽的电荷密度计算了硅的能带结构和电子态密度。
(一)优化晶格常数只需要优化一个晶格常数a。
首先准备四个文件:INCAR,POSCAR,POTCAR,KPOINTS,内容分别如下运行VASP,读取OUTCAR文件,可以得到a=5.1的情况下对应的能量。
然后改变a的值,再运行VASP,得到相应晶格常数对应的能量。
a的取值范围为5.1到5.8,每隔0.1取一个点。
最终得到晶格常数与能量的对应值如下表所示能量最小值为-10.83910312,对应的晶格常数为5.5,即为优化的晶格常数,后面的计算均基于此优化的晶格常数。
(二)静态计算自洽的电荷密度修改输入文件,在INCAR中定义NSW=0、LCHARG=T,POSCAR中的晶格常数定义为优化的晶格常数5.5,其它参数不变。
运行VASP,即得到了自洽的面电荷密度,将其保存下来,以便后面计算能带结构和电子态密度之用。
(三)计算能带结构选取6个特殊K点,特殊K点间的分割点数分别为20、20、20、10、20。
从自洽的电荷密度计算得到的OUTCAR文件中可以找到倒格子基矢和费米能级。
将以上信息写入syml文件,然后将syml文件输入到程序gk.x,产生K点,得到KPOINTS文件。
另外设置INCAR中的ISTART=1,ICHAGE=11,NSW=0。
POSCAR中的晶格常数定义为优化的晶格常数5.5。
其他参数不变。
运行VASP,计算完后得到本征值文件EIGENVAL。
输入文件EIGENVAL和syml到程序pbnf.x,可得到输出文件bnd.dat和highk。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
用vasp计算硅的能带结构
在最此次仿真之前,因为从未用过vasp软件,所以必须得学习此软件及一些能带的知识。
vasp是使用赝势和平面波基组,进行从头量子力学分子动力学计算的软件包。
用vasp计算硅的能带结构首先要了解晶体硅的结构,它是两个嵌套在一起的FCC布拉菲晶格,相对的位置为 (a/4,a/4,a/4), 其中a=5.4A是大的正方晶格的晶格常数。
在计算中,我们采用FCC的原胞,每个原胞里有两个硅原子。
VASP计算需要以下的四个文件:INCAR(控制参数), KPOINTS(倒空间撒点), POSCAR(原子坐标), POTCAR(赝势文件)
为了计算能带结构,我们首先要进行一次自洽计算,得到体系正确的基态电子密度。
然后固定此电荷分布,对于选定的特殊的K点进一步进行非自洽的能带计算。
有了需要的K点的能量本征值,也就得到了我们所需要的能带。
步骤一.—自洽计算产生正确的基态电子密度:
以下是用到的各个文件样本:
INCAR 文件:
SYSTEM = Si
Startparameter for this run:
NWRITE = 2; LPETIM=F write-flag & timer
PREC = medium medium, high low
ISTART = 0 job : 0-new 1-cont 2-samecut
ICHARG = 2 charge: 1-file 2-atom 10-const
ISPIN = 1 spin polarized calculation?
Electronic Relaxation 1
NELM = 90; NELMIN= 8; NELMDL= 10 # of ELM steps
EDIFF = 0.1E-03 stopping-criterion for ELM
LREAL = .FALSE. real-space projection
Ionic relaxation
EDIFFG = 0.1E-02 stopping-criterion for IOM
NSW = 0 number of steps for IOM
IBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CG
ISIF = 2 stress and relaxation
POTIM = 0.10 time-step for ionic-motion
TEIN = 0.0 initial temperature
TEBEG = 0.0; TEEND = 0.0 temperature during run
DOS related values:
ISMEAR = 0 ; SIGMA = 0.10 broadening in eV -4-tet -1-fermi 0-gaus
Electronic relaxation 2 (details)
Write flags
LWAVE = T write WAVECAR
LCHARG = T write CHGCAR
VASP给INCAR文件中的很多参数都设置了默认值,所以如果你对参数不熟悉,可以直接用默认的参数值。
比如在这个例子中,下面的比较简单的INCAR 文件也可以完成任务:
SYSTEM = Si
Startparameter for this run:
PREC = medium medium, high low
ISTART = 0 job : 0-new 1-cont 2-samecut
ICHARG = 2 charge: 1-file 2-atom 10-const
EDIFF = 0.1E-03 stopping-criterion for ELM
NSW = 0 number of steps for IOM
IBRION = 2 ionic relax: 0-MD 1-quasi-New 2-CG ISIF = 2 stress and relaxation
KPOINT文件:
我们采用自动的Monkhorst-Pack K点撒取方式。
对于类似于硅晶体的半导体材料,通常 4x4x4 的K点网格就够了。
Monkhorst Pack
Monkhorst Pack
4 4 4
0 0 0
POSCAR文件:
我们采用FCC原胞,所以每个原胞包含两个硅原子
Si
5.38936
0.5 0.5 0.0
0.0 0.5 0.5
0.5 0.0 0.5
2
Cartesian
0.0000000000000 0.00000000000 0.000000000000 0
0.2500000000000 0.25000000000 0.250000000000 0
POTCAR文件
不需要进行任何改动,只需将POTCAR文件从正确的赝势库里拷贝过来就行了。
运行VASP进行完这一步的计算后,我们应该得到了自洽的电荷分布-CHGCAR文件。
为了得到能带结构,我们需要对指定的K点进行非自洽的计算,然后将信息汇总,得到E-K的能带关系。
步骤二.—在固定电子密度的情况下,得到选取K点的能量本征值。
我们需要修改一下INCAR文件中的部分参数
ICHARG = 11 charge: 1-file 2-atom 10-const
ICHARG=11 表示从CHGCAR中读入电荷分布,并且在计算中保持不变。
我们还需要更改KPOINT文件,来指定我们感兴趣的某些高对称性的K点。
在VASP4.6中,这个可以通过Line mode来轻易实现.
k-points along high symmetry lines
10 ! 10 intersections
Line-mode
rec
0 0 0 ! gamma
0.5 0.5 0 ! X
0.0 0.0 0 ! gamma
0.5 0.5 0.5 ! L
通过指定Line-mode, VASP会自动在起点和终点之间插入指定的K点数,比如上面的文件就是指定VASP计算沿着Gamma点到X点,以及Gamma点到L点的K 点,每个方向上各取10个K点。
下图是硅晶体的第一布里渊区,标出了一些高对称性点。
作如上修改后,我们再次运行VASP,然后我们就可以从OUTCAR文件或者EIGENVAL 文件里得到需要的每个K点的能级信息。
比如说EIGENVAL文件会有类似以下的输出
0.5555556E-01 0.5555556E-01 0.0000000E+00 0.5000000E-01
1 -6.8356
2 4.8911
3 5.0077
4 5.0079
5 7.6438
6 8.0693
7 8.0694
8 9.0057
第一行就是K点的倒空间的坐标,接下来的8行告诉我们在那个K点上的8个能级。
你可以通过EXCEL或者ORIGIN之类的画图软件可视化结果。
由于现在手头上已经有了每个K点的能级信息,则将这些K点的能级连接起来就是你所需要的能带图了。
下图是用以上步骤算得的硅的能带图。
我们可以看到硅并非是直接能隙的材料。
同时,由于我们用了LDA,所以硅的能隙和实验相比大大被低估了(实验为1.12eV,计算值~0.6eV)。