肾上腺受体分子药理学
药理学考试重点
药理学考试重点1、受体、激动药、拮抗药、治疗指数概念受体:是一类介导细胞信号转导的功能蛋白质,能识别周围环境中某种微量化学物质,首先与之结合,并通过中介的信号放大系统,触发后续的生理反应或药理效应。
激动药:为既有亲和力又有内在活性的药物,能和受体结合并激动受体而产生效应。
拮抗药:能与受体结合,具有较强亲和力而无内在活性的药物。
治疗指数(TI):半数致死量和半数有效量的比值称为治疗指数。
治疗指数大的药物相对较治疗指数小的药物安全。
2、影响药物作用的主要因素(1)药物方面的因素:a.药物剂型:相同药物不同剂型,药物吸收速度和吸收的量可能不同,导致药物起效时间和作用强度的差异。
b.联合用药及药物相互作用:联合用药可能在药动学和药效学方面发生相互作用致药物作用改变。
(2)机体方面因素:年龄、性别、遗传、病理和心理因素对药物作用均可能产生影响。
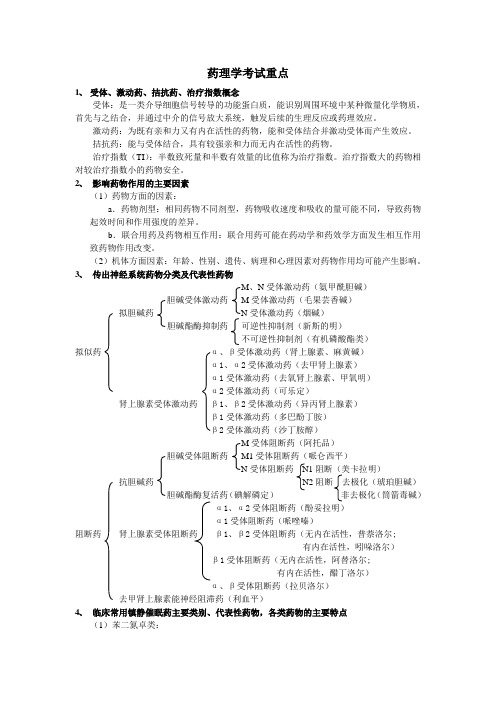
3、传出神经系统药物分类及代表性药物M、N受体激动药(氨甲酰胆碱)胆碱受体激动药M受体激动药(毛果芸香碱)拟胆碱药N受体激动药(烟碱)胆碱酯酶抑制药可逆性抑制剂(新斯的明)不可逆性抑制剂(有机磷酸酯类)拟似药α、β受体激动药(肾上腺素、麻黄碱)α1、α2受体激动药(去甲肾上腺素)α1受体激动药(去氧肾上腺素、甲氧明)α2受体激动药(可乐定)肾上腺素受体激动药β1、β2受体激动药(异丙肾上腺素)β1受体激动药(多巴酚丁胺)β2受体激动药(沙丁胺醇)M受体阻断药(阿托品)胆碱受体阻断药M1受体阻断药(哌仑西平)N受体阻断药N1阻断(美卡拉明)抗胆碱药N2阻断去极化(琥珀胆碱)胆碱酯酶复活药(碘解磷定)非去极化(筒箭毒碱)α1、α2受体阻断药(酚妥拉明)α1受体阻断药(哌唑嗪)阻断药肾上腺素受体阻断药β1、β2受体阻断药(无内在活性,普萘洛尔;有内在活性,吲哚洛尔)β1受体阻断药(无内在活性,阿替洛尔;有内在活性,醋丁洛尔)α、β受体阻断药(拉贝洛尔)去甲肾上腺素能神经阻滞药(利血平)4、临床常用镇静催眠药主要类别、代表性药物,各类药物的主要特点(1)苯二氮卓类:代表性药物有地西泮(安定)、三唑仑等,其特点是有较好的抗焦虑和镇静催眠作用,安全范围大。
药理学药物作用及其机制总结

药物药理作用及作用机制1 / 382 / 383 / 384 / 385 / 386 / 387 / 388 / 389 / 38疾病首选药物汇总10 / 38药品中毒解救11 / 38药物的主要不良反应及相似药物的比较12 / 3813 / 3814 / 38药物的主要临床用途15 / 3816 / 3817 / 3819 / 3820 / 38最严重的不良反应:硫脲类——粒细胞缺乏症;氯丙嗪——锥体外系反应;强心苷——心脏反应(如快速型心律失常、房室传导阻滞和窦性心动过缓)。
药理学口诀----天,居然有这个不可能不收藏啊拟胆碱药拟胆碱药分两类,兴奋受体抑制酶;匹罗卡品作用眼,外用治疗青光眼;新斯的明抗酯酶,主治重症肌无力;毒扁豆碱毒性大,作用眼科降眼压。
阿托品莨菪碱类阿托品,抑制腺体平滑肌;瞳孔扩大眼压升,调节麻痹心率快;大量改善微循环,中枢兴奋须防范;作用广泛有利弊,应用注意心血管。
21 / 38临床用途有六点,胃肠绞痛立即缓;抑制分泌麻醉前,散瞳配镜眼底检;防止“虹晶粘”,能治心动缓;感染休克解痉挛,有机磷中毒它首选。
东莨菪碱镇静显著东莨菪碱,能抗晕动是特点;可治哮喘和“震颤”,其余都像阿托品,只是不用它点眼。
肾上腺素α、β受体兴奋药,肾上腺素是代表;血管收缩血压升,局麻用它延时间,局部止血效明显,过敏休克当首选,心脏兴奋气管扩,哮喘持续它能缓,心跳骤停用“三联”,应用注意心血管,α受体被阻断,升压作用能翻转。
22 / 38去甲肾上腺素去甲强烈缩血管,升压作用不翻转,只能静滴要缓慢,引起肾衰很常见,用药期间看尿量,休克早用间羟胺。
异丙肾上腺素异丙扩张支气管,哮喘急发它能缓,扩张血管治“感染”,血容补足效才显。
兴奋心脏复心跳,加速传导律不乱,哮喘耐受防猝死,甲亢冠心切莫选。
α受体阻断药α受体阻断药,酚妥拉明酚苄明,扩张血管治栓塞,血压下降诊治瘤,NA释放心力增,治疗休克及心衰。
β受体阻断药23 / 38β受体阻断药,普萘洛尔是代表,临床治疗高血压,心律失常心绞痛。
β2肾上腺素能受体激动剂的应用

第一节概述β2-肾上腺素能受体激动剂(简称β2-受体激动剂)是目前临床应用较广、种类较多的支气管解痉剂,尤其是β2-受体激动剂的吸入剂型已广泛用于支气管哮喘的急性发作的治疗,可以有效地缓解哮喘的急性症状。
β2-受体激动剂具有很强的平喘作用,其支气管扩张效应是氨茶碱1000倍左右,因此在80年代以前的数10年里β2-受体激动剂一直作为支气管哮喘的首选药物而广泛使用,迄今仍是目前支气管哮喘急性期治疗的主要药物之一。
β2-肾上腺素能受体激动剂应用临床治疗哮喘已有近百年的历史,在本世纪初发现了包括麻黄素、肾上腺素、异丙基肾上腺素等β-肾上腺素能受体激动剂,这些β-肾上腺素能受体激动剂由于对β2-肾上腺素能受体选择性较差,具有较强的心血管副作用,目前已很少用于支气管哮喘的治疗,点介绍。
自60年代以来,具有选择性强、疗效好、副作用少的β2-受体激动剂逐渐进入临床,此后先后发现了30余种β2-受体激动剂,其中10余种已用于临床。
进入80年代后期,随着长效β2-受体激动剂的出现,使每日的用药的次数由过去的每日4~6次减为每日1~2次,尤其是配合吸入剂型给药,在临床上取得了很好的缓解哮喘症状的疗效。
同时由于这些长效β2-受体激动剂具有对β2-肾上腺素能受体有较强的选择性,大大降低了药物副作用发生的机率。
近几年许多药理学专家和哮喘病学专家从分子药理学水平对β2-受体激动剂治疗哮喘的作用机理进行了更加深入的研究,并取得了很大进展,也使β2受体激动剂的选择性更强,疗效更好和新的剂型不断问世,使β2-受体激动剂成为目前缓解哮喘急性症状的首选药物之一。
到目前为止,还没有发现β2-受体激动剂具有气遭抗炎效应,某些研究还证实单独应用β2-受体激动剂还可能加重气道炎症,这是由于β2 -受体激动剂仅仅是一种对症治疗的药物,其虽然可以暂时缓解哮喘症状,但易导致医生或病人忽视抗炎治疗,使气道炎症潜隐发展,加重气道高反应性,从而导致病情恶化,因此使用β2-受体激动剂的同时应配合其他抗炎治疗措施。
受体的概念及意义

受体的概念及意义受体是生物体中的一种特定分子结构,它能够与某些分子或化合物发生特异性的相互作用,从而导致信号传导、物质药效等生物效应的产生。
受体通常分布在细胞膜上,也可以存在于细胞质或细胞核内。
受体的概念主要起源于药理学和生物化学领域。
药理学角度来看,受体是指药物与生物体之间相互作用的特定靶点。
生物化学角度来看,受体是指与配体结合能够发生化学或物理变化的分子结构。
不同的生物体和不同的细胞具有不同的受体,这使得受体的研究成为药物设计和生理学研究的重要基础。
受体的意义主要体现在以下几个方面:1. 信号传导:细胞受体通过与配体结合,引起一系列内部信号转导的级联反应。
例如,肌肉细胞上的肾上腺素受体与外界的肾上腺素分子结合后,可以激活腺苷酸环化酶,从而产生环磷酸腺苷(cAMP),进而激活蛋白激酶A,最终导致肌肉收缩。
2. 药物作用:受体的发现和研究为药物设计和开发提供了重要的依据。
药物通常通过与受体结合或影响受体的活性来发挥作用。
药物的研发研究人员通过研究受体的结构和功能,可以更好地理解药物和受体之间的相互作用机制,从而设计出更加有效和有选择性的药物。
3. 生理调节:受体在生理活动中起到了重要的调节作用。
例如,许多内分泌系统靶标受体对于激素的抑制或促进起到了调节作用。
胰岛素受体对胰岛素的作用敏感,能够通过调节血糖水平来维持机体的内稳态;甲状腺受体对甲状腺激素的作用敏感,通过调节代谢率、能量平衡等来维持机体的正常功能。
4. 疾病诊断与治疗:许多疾病与受体的异常有关,如肿瘤、心血管疾病和神经系统疾病等。
研究受体的变化可以帮助我们了解疾病的发生和发展机制,并为疾病的早期诊断和治疗提供依据。
例如,癌症药物的设计常常针对特定的受体,如HER2受体在乳腺癌中的过度表达,通过设计针对HER2的抗体药物进行治疗。
受体的研究方法主要有两个方面:1. 结构研究,通过X射线晶体学、核磁共振等方法,解析受体的三维结构,从而揭示其与配体结合的机制;2. 功能研究,通过细胞生物学、分子生物学等方法,研究受体的功能,探究其在细胞内部信号传导及生理调控等方面的作用。
β肾上腺素受体在心血管系统的基础与临床研究进展(最全版)

β肾上腺素受体在心血管系统的基础与临床研究进展(最全版)β肾上腺素受体(β-adrenergic receptor,β-AR)是G蛋白偶联受体超家族(G protein-coupled receptors,GPCRs)的典型成员,主要在调节心血管系统活动中发挥作用,是心脏表达最丰富的受体。
β-AR信号系统的异常是众多心血管疾病发生发展的基础。
因此,在心血管系统中,对β-AR及β受体阻滞剂的研究一直是重中之重,其指导着人们不断优化心血管疾病的治疗效果。
1 β-AR的研究历史对β-AR的认识应追溯到受体学说提出之后,于1906年Dale首次引出了肾上腺素受体的概念;1948年Ahlquist基于拟交感药物在血管的不同反应又提出肾上腺素受体可分为α和β两种亚型;而后1962年,James[1]研究出了第一个β受体阻滞剂—普萘洛尔,被誉为20世纪药理学和药物治疗学上里程碑式的重大发现,并于1988年获得诺贝尔生理学或医学奖。
1967年Lands等根据受体药理学特性的不同,又进一步把β-AR分为β1-AR和β2-AR两种亚型;而基于此更深一层的认识和普萘洛尔发生的不良反应,人们又研发了以美托洛尔为代表的第二代选择性β1-AR阻滞剂,疗效和安全性都有很大提高。
此后,随着GPCR及信号转导通路的发现,β-AR作为GPCR的模式受体被不断深入研究,人们发现β-AR不仅偶联经典的G蛋白传导信号,还能偶联β-arrestin传递信号[2]。
β-arrestin的发现又推动β-AR的研究从针对受体层面进入到了针对β-AR下游信号转导分子及通路功能的时代,β-AR亚型信号通路、偏向激活、转位激活等更深层次的生理和病理性机制被不断发掘,新一代高效低毒的信号通路选择性药物也即将诞生。
2 β-AR的信号转导通路在G蛋白依赖性信号通路中,β-AR通过偶联G蛋白传递多种信号。
经典的信号通路遵循β-AR-G蛋白-环磷酸腺苷(cyclic adenosine monophosphate,cAMP)-蛋白激酶A(protein kinase A, PKA)-效应分子途径,如β1-AR激活后引起与之偶联的与Gs蛋白(激动亚型)解离出具有活性的Gα亚基,而后Gα的效应蛋白腺苷酸环化酶(adnylate cyclase, AC)催化三磷酸腺苷(adenosine triphosphate, ATP)生成cAMP。
药理学名词解释

药理学:研究药物与机体相互作用及其作用规律的一门学科药物效应动力学(药效学):研究药物对机体的作用极其作用的规律,也就是药物是如何起效的药物代谢动力学(药动学):主要是研究药物的吸收、分布、代谢、排泄的过程及其影响因素,并运用数学原理和方法阐释药物在体内的动态规律。
药物:用于预防、治疗、诊断及某些特殊用途的化学物质被称之为是药物古代药物(传统药物):是指以植物、动物或者是矿物质及其加工品入药。
现代药物:包括人工合成药物、天然药物的有效成分、生物制品以及基因工程药物。
药品:必须取得国家批准文号生产出来的合格的药物的成品才叫药品处方药:必须评职业医师的处方才可购买的药物。
包括强心苷类的药物、抗肿瘤的药物、刚上市的新药、所有的抗生素、注射剂等非处方药(OTC):不需要处方即可购买的药物。
分为甲类和乙类,甲类的标识为红底白字,乙类为绿底白字,乙类的不良反应更小。
新药:指我国未生产过的药品,已经生产过的药品改变剂型、改变给药途径、增加新的适应症或制成新的复方制剂。
药物作用:药物对机体的初始作用药物效应:是指药物作用所引起的机体生理、生化或者是形态的改变,是药物作用的结果。
兴奋作用:在药物的作用下,使机体原有的功能提高或是增强抑制作用:在药物的作用下,使机体原有的功能降低或是减弱直接作用:药物直接对它所接触的器官、组织、细胞所产生的作用间接作用:药物的直接作用进一步引起的作用药物选择性:是指在全身用药的情况下,药物对某些器官、组织有作用,而对另外一些器官或组织可能是没有用的药物的特异性:药物发挥作用需要和它的靶器官或者是靶部位进行结合,这种结合取决于药物和靶点的化学结构,特异性是药物对靶点的专一性。
局部作用:药物进入到血液循环之前,在给药部位发挥的作用。
全身作用:药物被吸收进入到血液循环之后分布到机体各个器官而产生的作用。
药物作用的两重性:药物在发挥作用的同时不可避免的会出现一些不良反应。
治疗作用:凡是符合用药目的,具有防治疾病效果的作用称为治疗作用,分为对因治疗和对症治疗对因治疗:消除原发致病因子的治疗对症治疗:改善基本症状的治疗替代疗法(补充疗法):临床使用药物补充体内营养或代谢物的缺乏,称为替代疗法。
药理学—名词解释

药物:指用于治疗、预防和诊断疾病的化学物质。
药理学:是研究药物与机体或病原体相互作用的规律和原理的一门学科。
药效动力学:是研究药物对机体的作用及作用机制的学科,以阐明药物防治疾病的规律。
药代动力学:是研究机体对药物处置的动态变化的学科。
包括药物在机体内的吸收、分布、生物转化(代谢)及排泄过程,特别是血药浓度随时间变化的规律。
一般药理学研究:指对新药主要药效作用以外的广泛药理作用研究。
主要是研究药物对精神、神经系统,心血管系统,呼吸系统以及其他系统的作用等。
安慰剂:是不含活性药物但又暗示某种效应的制剂(如在外形、颜色、味道等方面与某种活性药物或受试药物相同)药物作用:指药物与机体组织间的原发作用药物效应:药物原发作用所引起的机体器官原有功能的改变。
兴奋:凡能使机体生理、生化功能加强的作用称为..抑制:凡能引起功能活动减弱的作用受体:一类介导细胞信号转导的功能蛋白质,能识别周围环境中的某些微量化学物质,首先与之结合,并通过中介的信息放大系统,触发后续的生理反应或药理反应。
配体:能与受体特异性结合的物质副作用:指用于治疗量药物后出现的与治疗无关的不适反应慢性毒性:指长期用药而逐渐发生的毒性作用二重感染:长期使用四环素类等广谱性抗生素后,由于敏感菌株被抑制,而使肠道内菌群间的相对平衡被破坏,一些不敏感的细菌大量繁殖,而引起的继发性感染后遗效应:停药后血药浓度虽已降至最低有效浓度以下,但仍残存的生物效应部分激动剂:与受体有亲和力,但内在活性低的药物。
竞争性拮抗剂:与受体有亲和力但没有内在活性,且激动剂相互竞争相同的受体极量:产生疗效的最大治疗量药物的效能:指药物引起的最大效应效价:指产生相同效应时所需要剂量或浓度大小,与药物的作用强度成反比治疗指数:是LD50与ED50的比值,该指数越大表示药物越安全首关效应:指口服药物在胃肠道吸收后,首先进入肝门静脉系统,某些药物在通过肠黏膜及肝脏时,部分可被代谢灭活而进入体循环的药量减少,药效降低表观分布容积:当药物在体内分布达到动态平衡时,体内药量与血药浓度的比值称...消除半衰期:血药浓度降低一半所需要的时间个体差异:在相同用药情况下,不同个体对药物的反应有所不同快速耐受性:在短时间内连续用药数次后,立即产生的耐受性交叉耐受性:机体对某些产生耐受性后,对另一药的敏感也降低特异质:少数病人对某些药物出现极敏感或极不敏感的反应,或出现与通常性质不同的反应躯体依赖性:反复用药后造成身体适应状态产生欣快感,一旦中断用药,可出现强烈的戒断综合症,称为精神依赖性:用药后产生愉快满足的感觉,使用药者在精神上渴望周期性或连续用药,以达到舒适感药物相互作用:两种或多种药物合用或先后序贯应用,而引起药物作用和效应的变化,称.. 协同作用:合并用药作用增加总称...拮抗作用:合并用药效应减弱,两药合用的效应小于它们分别作用的总和胆碱受体:能与ACh结合的受体。
_2肾上腺素受体的结构及其生物学功能

马旁回、扣带回等处密度最高。在蓝斑、脑干 A4、A5、A7 区仅有 α2A- AR 亚型分布[4]。
α2- AR 由 450 个 氨 基 酸 残 基 组 成 , 为 7 次 跨 膜受体, 含有多个亲水区与疏水区, 其疏水区迂 回 进 出 细 胞 膜 , 由 20~25 个 氨 基 酸 残 基 组 成 的 α 螺旋结构组成。其亲水区包括 3 个胞外环和 3 个 胞内环。
临床药物 的 开 发 有 着 极 其 重 要 的 作 用 , α2- AR 亚 型作为靶标是筛选高选择性和高活性药物的理想 工具。 1. α2- AR 的分型与命名
肾上腺素受体是指与去甲肾上腺素和肾上腺 素结合的受体总称。根据肾上腺素受体药理学特 征, 分为 α和 β两大类。
随着选择性配基的出现, α肾上腺素受体又可 分 为 α1 和 α2 两 种 受 体 亚 型 , 其 中 α2 受 体 亚 型 , 在人类及其他哺乳动物中又可表达为三种亚型, 分别为 α2A (α2D)、α2B 和 α2C。α2A、α2B 和 α2C 的基因 依 次 定 位 于 10、2 和 4 号 染 色 体 上 , 基 因 命 名 为 α2C10、α2C2 和 α2C4[2]。这 3 个 α2- AR 亚型的重要 结构与功能区域是较为保守的, 但各亚型间氨基 酸 同 源 性 仅 50%。 啮 齿 类 动 物 中 RG20 基 因 与 人 α2C10 基因高度同源(84%), RG20 基因编码的 α2D- AR 与 α2A- AR 氨基酸序列的同源性 高 达 89%, 但 二者的药理学特性存在差异, 表明 α2D- AR 是 α2A- AR 的种属变体, 故未被列为 α2- AR 的亚型[3]。 2. α2- AR 的分子结构特点
目前, 国内部分科研机构已成功从乳猪或乳 牛 肝 脏 中 提 取 出 了 较 为 纯 化 的 HSS, 并 获 得 了 国 家新药证书, 有注射液、口服液等多种形式, 已 广泛应用于临床, 对重症型肝炎、急性肝炎、慢 性肝炎、肝硬化等均有良好的疗效。由于我国是 肝炎大国, 而且 HSS 的原料来源广泛, 制作流程 较为简单, 制品可以批量生产, 冻干保存, 所以 有非常理想的应用前景。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第一节 肾上腺素受体的分型
分型的标准: ①对特异性配体(包括拮抗剂及激动剂)
的亲和性 ②激动后信号传导机制及生物学效应的
特点 ③基因的结构以及在染色体上的位置
• 表1-1 α1-AR,α2-AR及β-AR的药理及信 号传导系统特征
α1-AR的亚型
• Morrow及Greese于1986年用3H-哌唑嗪作 为放射性配体显示拮抗剂WB4101及酚妥 拉明及大鼠脑皮层中的α1-AR有高、低亲 和性两种结合位点,假设它们为α1-AR的 2种亚型,并分别称之为α1A及α1B亚型 。
• ②点突变(site mutation)法:即有目的地 改变基因DNA中的碱基对序列由别的氨 基酸来代替;
• ③嵌合(Chimeric)法:即在两种亚型AR间 交换某一区域结构,嵌合成一种新的包 含两种亚型结构的受体。
细胞外区域的功能
• 糖基化位点:其相连的糖起什么作用至 今不明,至少可以肯定它们及配体结合 特性没有关系。
• 随后不久也发现脂肪组织中部分β-AR的 药理特性及βl-AR、β2-AR均不相同
β-AR的亚型(con’t)
• 1989年Emorine等采用火鸡β1-Aβ2AR的克隆基因
• 其编码402个氨基酸,序列及人β1-AR及 β2-AR分别仅有50.7%和45.5%相同(人 β1-AR及β2-AR之间有48.9%相同),称之 为β3-AR。
• 信号传导机制:α1A-AR激动后引起的生物学 效应依赖于细胞外Ca2+的存在,而α1B-AR不 依赖。2种亚型受体激动时生成磷酸肌醇的种 类、生成途径及时相等都有明显差别。
关于α1c-AR亚型
• 1990年Schwinn等从牛脑得到一种新的α1-AR cDNA克隆,其中跨膜部位氨基酸序列有72% 及α1B-AR相同。
AR分子的基本结构
• AR分子由429-561个氨基酸残基组成,有 7个区域,为连续20-28个疏水氨基酸组 成,形成7个跨膜片段
• 构成3个细胞外环及3个细胞内环,受体 分子的氨基端在胞外,羧基端在胞内。
AR结构示意图
研究AR结构及功能的方法
• ①删除(deletion)法:即有目的地去除一 段基因DNA,使表达的受体缺少一段氨 基酸序列;
B亚型受体的药理特性 及信号传导系统的主要差别
• 及氯乙基可乐定(CEC)的反应:仅α1B-AR及 CEC发生烷化反应从而被不可逆阻断
• 及选择性拮抗剂及激动剂的亲和性:及多数拮 抗剂如WB4l01、酚妥拉明等及激动剂甲氧明、 部分激动剂羟甲唑啉等的亲和性α1A-AR显著 高于α1B-AR,仅对拮抗剂Spiperone的亲和性 为α1B-AR高于α1A-AR
• 在COS7细胞表达后检查这种受体的药理特性 ,发现它及WB4101、酚妥拉明等的亲和性显 著高于α1B-AR,而及α1A-AR相近,但却又能 被CEC不可逆性阻断,因而既不同于α1A-AR ,又不同于α1B-AR,他们称这种受体为α1C亚 型。
关于α1c-AR亚型(con’t)
• Faure等、Rokosh等、price等、Perez等陆 续采用更为精确的RNase保护分析方法显 示大鼠α1C克隆受体在大鼠组织中的分布 及α1A亚型相同。
α2-AR的亚型
• 根据对选择性拮抗剂的亲和性及组织分 布特异性,可将α2-AR分成α2A、α2B及 α2C 3种亚型
• 见表1-3 α2-AR各种亚型的药理学特性
采用分子生物学技术的分型
• 最先分别从人血小板、基因库及肾脏克 隆到3种α2-AR,其基因分别位于第10、 第2及第4对染色体上,故称作α2-C10、 α2-C2及α2-C4亚型
• Ford等及Blue等还显示大鼠α1C克隆受体 的药理特性及α1A亚型相似。
• 因此,国际药理联合会受体命名及药物 分类委员会决定取消α1C而将原来的改为 α1A
α1D亚型 的发现
• 1991年 Perez等从大鼠脑海马回中克隆到一种 α1-AR cDNA,它的碱基对序列及Lomasney报 道的克隆只差2个碱基对
• α2-C10的药理特性及分布及α2A完全相符 。关于α2-C2、α2-C10及α2B、α2C间的 关系尚未完全定论,但多数意见认为α2C2及α2B对应,α2-C4及α2C对应。
β-AR的亚型
• 60年代中后期已确定β-AR包含β1及β2亚 型,
• 及内源性激动剂的亲和性:β1-AR为 EPi≤NE,而在β2-AR则Epi>NE
• 在COS7细胞表达后的药理学特性检查,发现 它及WB4101及5-MU的亲和性虽略高于α1BAR,但又显著低于α1A-AR
• 对(+)niguldipine的亲和性甚至比α1B亚型还低 ,而且可部分被CEC不可逆阻断。因而他们认 为这是第四种α1-AR亚型,称之为α1D。
表1-2 α1-AR三种亚型的主要区 别
β3-AR及βl-AR、β2-AR的药理 特性
• 将人β3-AR基因及人βl-AR、人β3-AR cDNA分别在CHO细胞表达后,观察及比 较这三种β-AR亚型的药理特性发现β3AR及βl-AR、β2-AR显著不同,而及以前 报道的非典型β-AR基本一致(见表1-4)
第二节 肾上腺素受体结构及功 能的关系
• ②在β2-AR删除亲水性细胞外环或细胞内 环各区域片段,对配体结合特点并无影 响,但当删除疏水性跨膜片段的氨基酸 片段时,则见配体结合能力明显减弱。 如改换β2-AR第Ⅰ、第Ⅱ、第Ⅲ或第Ⅶ跨 膜区中若干高度保守氨基酸中任意一个 氨基酸,其配体结合性质也发生重大改 变;
• 细胞外环中的某些半胱氨酸可通过二硫 键来稳定配体结合袋(Ligand-binding pocket)。用其他氨基酸取代其中一个半 胱氨酸都能使受体配体结合能力降低
跨膜区域的功能
• 7个跨膜单位在膜上按一定位置构成配体 结合袋。
• 证据有3个方面 :
• ①采用放射性光亲和性探针及α2-AR或 β2-AR共价结合,然后酶解AR蛋白质, 显示探针及AR共价结合的部位都在跨膜 区域