Kenichi Yoshida et al.,2012——结合癌症研究文献无重点介绍了几种测
Cancer Epigenetics From Mechanism to Therapy

Leading EdgeReviewCancer Epigenetics:From Mechanism to TherapyMark A.Dawson1,2and Tony Kouzarides1,*1Gurdon Institute and Department of Pathology,University of Cambridge,Tennis Court Road,Cambridge CB21QN,UK2Department of Haematology,Cambridge Institute for Medical Research and Addenbrooke’s Hospital,University of Cambridge,Hills Road, Cambridge CB20XY,UK*Correspondence:t.kouzarides@/10.1016/j.cell.2012.06.013The epigenetic regulation of DNA-templated processes has been intensely studied over the last15 years.DNA methylation,histone modification,nucleosome remodeling,and RNA-mediated target-ing regulate many biological processes that are fundamental to the genesis of cancer.Here,we present the basic principles behind these epigenetic pathways and highlight the evidence suggest-ing that their misregulation can culminate in cancer.This information,along with the promising clin-ical and preclinical results seen with epigenetic drugs against chromatin regulators,signifies that it is time to embrace the central role of epigenetics in cancer.Chromatin is the macromolecular complex of DNA and histone proteins,which provides the scaffold for the packaging of our entire genome.It contains the heritable material of eukaryotic cells.The basic functional unit of chromatin is the nucleosome. It contains147base pairs of DNA,which is wrapped around a histone octamer,with two each of histones H2A,H2B,H3, and H4.In general and simple terms,chromatin can be subdi-vided into two major regions:(1)heterochromatin,which is highly condensed,late to replicate,and primarily contains inac-tive genes;and(2)euchromatin,which is relatively open and contains most of the active genes.Efforts to study the coordi-nated regulation of the nucleosome have demonstrated that all of its components are subject to covalent modification,which fundamentally alters the organization and function of these basic tenants of chromatin(Allis et al.,2007).The term‘‘epigenetics’’was originally coined by Conrad Wad-dington to describe heritable changes in a cellular phenotype that were independent of alterations in the DNA sequence. Despite decades of debate and research,a consensus definition of epigenetics remains both contentious and ambiguous(Berger et al.,2009).Epigenetics is most commonly used to describe chromatin-based events that regulate DNA-templated pro-cesses,and this will be the definition we use in this review. Modifications to DNA and histones are dynamically laid down and removed by chromatin-modifying enzymes in a highly regulated manner.There are now at least four different DNA modifications(Baylin and Jones,2011;Wu and Zhang,2011) and16classes of histone modifications(Kouzarides,2007;Tan et al.,2011).These are described in Table1.These modifications can alter chromatin structure by altering noncovalent interac-tions within and between nucleosomes.They also serve as docking sites for specialized proteins with unique domains that specifically recognize these modifications.These chromatin readers recruit additional chromatin modifiers and remodeling enzymes,which serve as the effectors of the modification.The information conveyed by epigenetic modifications plays a critical role in the regulation of all DNA-based processes, such as transcription,DNA repair,and replication.Conse-quently,abnormal expression patterns or genomic alterations in chromatin regulators can have profound results and can lead to the induction and maintenance of various cancers.In this Review,we highlight recent advances in our understanding of these epigenetic pathways and discuss their role in oncogen-esis.We provide a comprehensive list of all the recurrent cancer mutations described thus far in epigenetic pathways regulating modifications of DNA(Figure2),histones(Figures3,4,and5), and chromatin remodeling(Figure6).Where relevant,we will also emphasize existing and emerging drug therapies aimed at targeting epigenetic regulators(Figure1).Characterizing the EpigenomeOur appreciation of epigenetic complexity and plasticity has dramatically increased over the last few years following the development of several global proteomic and genomic technol-ogies.The coupling of next-generation sequencing(NGS)plat-forms with established chromatin techniques such as chromatin immunoprecipitation(ChIP-Seq)has presented us with a previ-ously unparalleled view of the epigenome(Park,2009).These technologies have provided comprehensive maps of nucleo-some positioning(Segal and Widom,2009),chromatin confor-mation(de Wit and de Laat,2012),transcription factor binding sites(Farnham,2009),and the localization of histone(Rando and Chang,2009)and DNA(Laird,2010)modifications.In addi-tion,NGS has revealed surprising facts about the mammalian transcriptome.We now have a greater appreciation of the fact that most of our genome is transcribed and that noncoding RNA may play a fundamental role in epigenetic regulation(Ama-ral et al.,2008).Most of the complexity surrounding the epigenome comes from the modification pathways that have been identified.12Cell150,July6,2012ª2012Elsevier Inc.Recent improvements in the sensitivity and accuracy of mass spectrometry (MS)instruments have driven many of these discoveries (Stunnenberg and Vermeulen,2011).Moreover,although MS is inherently not quantitative,recent advances in labeling methodologies,such as stable isotope labeling by amino acids in cell culture (SILAC),isobaric tags for relative and absolute quantification (iTRAQ),and isotope-coded affinity tag (ICAT),have allowed a greater ability to provide quantitative measurements (Stunnenberg and Vermeulen,2011).These quantitative methods have generated ‘‘protein recruit-ment maps’’for histone and DNA modifications,which contain proteins that recognize chromatin modifications (Bartke et al.,2010;Vermeulen et al.,2010).Many of these chromatin readers have more than one reading motif,so it is important to under-stand how they recognize several modifications either simulta-neously or sequentially.The concept of multivalent engagement by chromatin-binding modules has recently been explored by using either modified histone peptides (Vermeulen et al.,2010)or in-vitro-assembled and -modified nucleosomes (Bartkeet al.,2010;Ruthenburg et al.,2011).The latter approach in particular has uncovered some of the rules governing the recruit-ment of protein complexes to methylated DNA and modified histones in a nucleosomal context.The next step in our under-standing will require a high-resolution in vivo genomic approach to detail the dynamic events on any given nucleosome during the course of gene expression.Epigenetics and the Cancer ConnectionThe earliest indications of an epigenetic link to cancer were derived from gene expression and DNA methylation studies.These studies are too numerous to comprehensively detail in this review;however,the reader is referred to an excellent review detailing the history of cancer epigenetics (Feinberg and Tycko,2004).Although many of these initial studies were purely correl-ative,they did highlight a potential connection between epige-netic pathways and cancer.These early observations have been significantly strengthened by recent results from the Inter-national Cancer Genome Consortium (ICGC).Whole-genomeTable 1.Chromatin Modifications,Readers,and Their Function Chromatin Modification NomenclatureChromatin-Reader MotifAttributed Functionand Cit,citrulline.Reader domains:MBD,methyl-CpG-binding domain;PHD,plant homeodomain;MBT,malignant brain tumor domain;PWWP,proline-tryptophan-tryptophan-proline domain;BRCT,BRCA1C terminus domain;UIM,ubiquitin interaction motif;IUIM,inverted ubiquitin interaction motif;SIM,sumo interaction motif;and PBZ,poly ADP-ribose binding zinc finger.aThese are established binding modules for the posttranslational modification;however,binding to modified histones has not been firmly established.Cell 150,July 6,2012ª2012Elsevier Inc.13sequencing in a vast array of cancers has provided a catalog of recurrent somatic mutations in numerous epigenetic regulators (Forbes et al.,2011;Stratton et al.,2009).A central tenet in analyzing these cancer genomes is the identification of ‘‘driver’’mutations (causally implicated in the process of oncogenesis).A key feature of driver mutations is that they are recurrently found in a variety of cancers,and/or they are often present at a high prevalence in a specific tumor type.We will mostly concentrate our discussions on suspected or proven driver mutations in epigenetic regulators.For instance,malignancies such as follicular lymphoma contain recurrent mutations of the histone methyltransferase MLL2in close to 90%of cases (Morin et al.,2011).Similarly,UTX ,a histone demethylase,is mutated in up to 12histologi-cally distinct cancers (van Haaften et al.,2009).Compilation of the epigenetic regulators mutated in cancer highlights histone acetylation and methylation as the most widely affected epige-netic pathways (Figures 3and 4).These and other pathways that are affected to a lesser extent will be described in the following sections.Deep sequencing technologies aimed at mapping chromatin modifications have also begun to shed some light on the origins of epigenetic abnormalities in cancer.Cross-referencing of DNA methylation profiles in human cancers with ChIP-Seq data for histone modifications and the binding of chromatinregulators have raised intriguing correlations between cancer-associated DNA hypermethylation and genes marked with ‘‘bivalent’’histone modifications in multipotent cells (Easwaran et al.,2012;Ohm et al.,2007).These bivalent genes are marked by active (H3K4me3)and repressive (H3K27me3)histone modi-fications (Bernstein et al.,2006)and appear to identify transcrip-tionally poised genes that are integral to development and lineage commitment.Interestingly,many of these genes are targeted for DNA methylation in cancer.Equally intriguing are recent comparisons between malignant and normal tissues from the same individuals.These data demonstrate broad domains within the malignant cells that contain significant alter-ations in DNA methylation.These regions appear to correlate with late-replicating regions of the genome associated with the nuclear lamina (Berman et al.,2012).Although there remains little mechanistic insight into how and why these regions of the genome are vulnerable to epigenetic alterations in cancer,these studies highlight the means by which global sequencing plat-forms have started to uncover avenues for further investigation.Genetic lesions in chromatin modifiers and global alterations in the epigenetic landscape not only imply a causative role for these proteins in cancer but also provide potential targets for therapeutic intervention.A number of small-molecule inhibitors have already been developed against chromatin regulators (Figure 1).These are at various stages of development,andthreeFigure 1.Epigenetic Inhibitors as Cancer TherapiesThis schematic depicts the process for epigenetic drug development and the current status of various epigenetic therapies.Candidate small molecules are first tested in vitro in malignant cell lines for specificity and phenotypic response.These may,in the first instance,assess the inhibition of proliferation,induction of apoptosis,or cell-cycle arrest.These phenotypic assays are often coupled to genomic and proteomic methods to identify potential molecular mechanisms for the observed response.Inhibitors that demonstrate potential in vitro are then tested in vivo in animal models of cancer to ascertain whether they may provide therapeutic benefit in terms of survival.Animal studies also provide valuable information regarding the toxicity and pharmacokinetic properties of the drug.Based on these preclinical studies,candidate molecules may be taken forward into the clinical setting.When new drugs prove beneficial in well-conducted clinical trials,they are approved for routine clinical use by regulatory authorities such as the FDA.KAT,histone lysine acetyltransferase;KMT,histone lysine methyltransferase;RMT,histone arginine methyltransferase;and PARP,poly ADP ribose polymerase.14Cell 150,July 6,2012ª2012Elsevier Inc.of these(targeting DNMTs,HDACs,and JAK2)have already been granted approval by the US Food and Drug Administra-tion(FDA).This success may suggest that the interest in epige-netic pathways as targets for drug discovery had been high over the past decade.However,the reality is that thefield of drug discovery had been somewhat held back due to concerns over the pleiotropic effects of both the drugs and their targets. Indeed,some of the approved drugs(against HDACs)have little enzyme specificity,and their mechanism of action remains contentious(Minucci and Pelicci,2006).The belief and investment in epigenetic cancer therapies may now gain momentum and reach a new level of support following the recent preclinical success of inhibitors against BRD4,an acetyl-lysine chromatin-binding protein(Dawson et al.,2011; Delmore et al.,2011;Filippakopoulos et al.,2010;Mertz et al., 2011;Zuber et al.,2011).The molecular mechanisms governing these impressive preclinical results have also been largely uncovered and are discussed below.This process is pivotal for the successful progression of these inhibitors into the clinic. These results,along with the growing list of genetic lesions in epigenetic regulators,highlight the fact that we have now entered an era of epigenetic cancer therapies.Epigenetic Pathways Connected to CancerDNA MethylationThe methylation of the5-carbon on cytosine residues(5mC)in CpG dinucleotides was thefirst described covalent modifica-tion of DNA and is perhaps the most extensively characterized modification of chromatin.DNA methylation is primarily noted within centromeres,telomeres,inactive X-chromosomes,and repeat sequences(Baylin and Jones,2011;Robertson,2005). Although global hypomethylation is commonly observed in malignant cells,the best-studied epigenetic alterations in cancerare the methylation changes that occur within CpG islands, which are present in 70%of all mammalian promoters.CpG island methylation plays an important role in transcriptional regu-lation,and it is commonly altered during malignant transforma-tion(Baylin and Jones,2011;Robertson,2005).NGS platforms have now provided genome-wide maps of CpG methylation. These have confirmed that between5%–10%of normally unme-thylated CpG promoter islands become abnormally methylated in various cancer genomes.They also demonstrate that CpG hypermethylation of promoters not only affects the expression of protein coding genes but also the expression of various noncoding RNAs,some of which have a role in malignant trans-formation(Baylin and Jones,2011).Importantly,these genome-wide DNA methylome studies have also uncovered intriguing alterations in DNA methylation within gene bodies and at CpG‘‘shores,’’which are conserved sequences upstream and downstream of CpG islands.The functional relevance of these regional alterations in methylation are yet to be fully deciphered, but it is interesting to note that they have challenged the general dogma that DNA methylation invariably equates with transcriptional silencing.In fact,these studies have established that many actively transcribed genes have high levels of DNA methylation within the gene body,suggesting that the context and spatial distribution of DNA methylation is vital in transcrip-tional regulation(Baylin and Jones,2011).Three active DNA methyltransferases(DNMTs)have been identified in higher eukaryotes.DNMT1is a maintenance methyl-transferase that recognizes hemimethylated DNA generated during DNA replication and then methylates newly synthesized CpG dinucleotides,whose partners on the parental strand are already methylated(Li et al.,1992).Conversely,DNMT3a and DNMT3b,although also capable of methylating hemimethylated DNA,function primarily as de novo methyltransferases to estab-lish DNA methylation during embryogenesis(Okano et al.,1999). DNA methylation provides a platform for several methyl-binding proteins.These include MBD1,MBD2,MBD3,and MeCP2. These in turn function to recruit histone-modifying enzymes to coordinate the chromatin-templated processes(Klose and Bird,2006).Although mutations in DNA methyltransferases and MBD proteins have long been known to contribute to developmental abnormalities(Robertson,2005),we have only recently become aware of somatic mutations of these key genes in human malig-nancies(Figure2).Recent sequencing of cancer genomes has identified recurrent mutations in DNMT3A in up to25%of patients with acute myeloid leukemia(AML)(Ley et al.,2010). Importantly,these mutations are invariably heterozygous and are predicted to disrupt the catalytic activity of the enzyme. Moreover,their presence appears to impact prognosis(Patel et al.,2012).However,at present,the mechanisms bywhich Figure2.Cancer Mutations Affecting Epigenetic Regulators of DNA MethylationThe5-carbon of cytosine nucleotides are methylated(5mC)by a family of DNMTs.One of these,DNMT3A,is mutated in AML,myeloproliferative diseases(MPD),and myelodysplastic syndromes(MDS).In addition to its catalytic activity,DNMT3A has a chromatin-reader motif,the PWWP domain, which may aid in localizing this enzyme to chromatin.Somatically acquired mutations in cancer may also affect this domain.The TET family of DNA hydroxylases metabolizes5mC into several oxidative intermediates,including 5-hydroxymethylcytosine(5hmC),5-formylcytosine(5fC),and5-carbox-ylcytosine(5caC).These intermediates are likely involved in the process of active DNA demethylation.Two of the three TET family members are mutated in cancers,including AML,MPD,MDS,and CMML.Mutation types are as follows:M,missense;F,frameshift;N,nonsense;S,splice site mutation;and T,translocation.Cell150,July6,2012ª2012Elsevier Inc.15these mutations contribute to the development and/or mainte-nance of AML remains elusive.Understanding the cellular consequences of normal and aber-rant DNA methylation remains a key area of interest,especially because hypomethylating agents are one of the few epigenetic therapies that have gained FDA approval for routine clinical use(Figure1).Although hypomethylating agents such as azaci-tidine and decitabine have shown mixed results in various solid malignancies,they have found a therapeutic niche in the myelo-dysplastic syndromes(MDS).Until recently,this group of disor-ders was largely refractory to therapeutic intervention,and MDS was primarily managed with supportive care.However,several large studies have now shown that treatment with azacitidine, even in poor prognosis patients,improves their quality of life and extends survival time.Indeed,azacitidine is thefirst therapy to have demonstrated a survival benefit for patients with MDS (Fenaux et al.,2009).The molecular mechanisms governing the impressive responses seen in MDS are largely unknown. However,recent evidence would suggest that low doses of these agents hold the key to therapeutic benefit(Tsai et al., 2012).It is also emerging that the combinatorial use of DNMT and HDAC inhibitors may offer superior therapeutic outcomes (Gore,2011).DNA Hydroxy-Methylation and Its Oxidation Derivatives Historically,DNA methylation was generally considered to be a relatively stable chromatin modification.However,early studies assessing the global distribution of this modification during embryogenesis had clearly identified an active global loss of DNA methylation in the early zygote,especially in the male pronucleus.More recently,high-resolution genome-wide mapping of this modification in pluripotent and differentiated cells has also confirmed the dynamic nature of DNA methylation, evidently signifying the existence of an enzymatic activity within mammalian cells that either erases or alters this chromatin modification(Baylin and Jones,2011).In2009,two seminal manuscripts describing the presence of5-hydroxymethylcyto-sine(5hmC)offered thefirst insights into the metabolism of 5mC(Kriaucionis and Heintz,2009;Tahiliani et al.,2009).The ten-eleven translocation(TET1–3)family of proteins have now been demonstrated to be the mammalian DNA hydroxy-lases responsible for catalytically converting5mC to5hmC. Indeed,iterative oxidation of5hmC by the TET family results in further oxidation derivatives,including5-formylcytosine(5fC) and5-carboxylcytosine(5caC).Although the biological signifi-cance of the5mC oxidation derivatives is yet to be established, several lines of evidence highlight their importance in transcrip-tional regulation:(1)they are likely to be an essential intermediate in the process of both active and passive DNA demethylation,(2) they preclude or enhance the binding of several MBD proteins and,as such,will have local and global effects by altering the recruitment of chromatin regulators,and(3)genome-wide mapping of5hmC has identified a distinctive distribution of this modification at both active and repressed genes,including its presence within gene bodies and at the promoters of bivalently marked,transcriptionally poised genes(Wu and Zhang,2011). Notably,5hmC was also mapped to several intergenic cis-regu-latory elements that are either functional enhancers or insulator elements.Consistent with the notion that5hmC is likely to have a role in both transcriptional activation and silencing, the TET proteins have also been shown to have activating and repressive functions(Wu and Zhang,2011).Genome-wide mapping of TET1has demonstrated it to have a strong prefer-ence for CpG-rich DNA and,consistent with its catalytic function, it also been localized to regions enriched for5mC and5hmC. The TET family of proteins derive their name from the initial description of a recurrent chromosomal translocation, t(10;11)(q22;q23),which juxtaposes the MLL gene with TET1in a subset of patients with AML(Lorsbach et al.,2003).Notably, concurrent to the initial description of the catalytic activity for the TET family of DNA hydroxylases,several reports emerged describing recurrent mutations in TET2in numerous hematolog-ical malignancies(Cimmino et al.,2011;Delhommeau et al., 2009;Langemeijer et al.,2009)(Figure2).Interestingly,TET2-deficient mice develop a chronic myelomonocytic leukemia (CMML)phenotype,which is in keeping with the high prevalence of TET2mutations in patients with this disease(Moran-Crusio et al.,2011;Quivoron et al.,2011).The clinical implications of TET2mutations have largely been inconclusive;however,in some subsets of AML patients,TET2mutations appear to confer a poor prognosis(Patel et al.,2012).Early insights into the process of TET2-mediated oncogenesis have revealed that the patient-associated mutations are largely loss-of-function muta-tions that consequently result in decreased5hmC levels and a reciprocal increase in5mC levels within the malignant cells that harbor them.Moreover,mutations in TET2also appear to confer enhanced self-renewal properties to the malignant clones (Cimmino et al.,2011).Histone ModificationsIn1964,Vincent Allfrey prophetically surmised that histone modifications might have a functional influence on the regulation of transcription(Allfrey et al.,1964).Nearly half a century later, thefield is still grappling with the task of unraveling the mecha-nisms underlying his enlightened statement.In this time,we have learned that these modifications have a major influence, not just on transcription,but in all DNA-templated processes (Kouzarides,2007).The major cellular processes attributed to each of these modifications are summarized in Table1.The great diversity in histone modifications introduces a remarkable complexity that is slowly beginning to be ing transcription as an example,we have learned that multiple coexisting histone modifications are associated with activation,and some are associated with repression. However,these modification patterns are not static entities but a dynamically changing and complex landscape that evolves in a cell context-dependent fashion.Moreover,active and repres-sive modifications are not always mutually exclusive,as evi-denced by‘‘bivalent domains.’’The combinatorial influence that one or more histone modifications have on the deposition, interpretation,or erasure of other histone modifications has been broadly termed‘‘histone crosstalk,’’and recent evidence would suggest that crosstalk is widespread and is of great bio-logical significance(Lee et al.,2010).It should be noted that the cellular enzymes that modify histones may also have nonhistone targets and,as such,it has been difficult to divorce the cellular consequences of individual histone modifications from the broader targets of many of these16Cell150,July6,2012ª2012Elsevier Inc.enzymes.In addition to their catalytic function,many chromatin modifiers also possess‘‘reader’’domains allowing them to bind to specific regions of the genome and respond to information conveyed by upstream signaling cascades.This is important, as it provides two avenues for therapeutically targeting these epigenetic regulators.The residues that line the binding pocket of reader domains can dictate a particular preference for specific modification states,whereas residues outside the binding pocket contribute to determining the histone sequence specificity.This combination allows similar reader domains to dock at different modified residues or at the same amino acid displaying different modification states.For example,some methyl-lysine readers engage most efficiently with di/tri-methyl-ated lysine(Kme2/3),whereas others prefer mono-or unmethy-lated lysines.Alternatively,when the same lysines are now acet-ylated,they bind to proteins containing bromodomains(Taverna et al.,2007).The main modification binding pockets contained within chromatin-associated proteins is summarized in Table1. Many of the proteins that modify or bind these histone modifi-cations are misregulated in cancer,and in the ensuing sections, we will discuss the most extensively studied histone modifica-tions in relation to oncogenesis and novel therapeutics. Histone Acetylation.The Nε-acetylation of lysine residues is a major histone modification involved in transcription,chromatin structure,and DNA repair.Acetylation neutralizes lysine’s posi-tive charge and may consequently weaken the electrostatic interaction between histones and negatively charged DNA.For this reason,histone acetylation is often associated with a more ‘‘open’’chromatin conformation.Consistent with this,ChIP-Seq analyses have confirmed the distribution of histone acetyla-tion at promoters and enhancers and,in some cases,throughout the transcribed region of active genes(Heintzman et al.,2007; Wang et al.,2008).Importantly,lysine acetylation also serves as the nidus for the binding of various proteins with bromodo-mains and tandem plant homeodomain(PHD)fingers,which recognize this modification(Taverna et al.,2007).Acetylation is highly dynamic and is regulated by the competing activities of two enzymatic families,the histone lysine acetyltransferases(KATs)and the histone deacetylases (HDACs).There are two major classes of KATs:(1)type-B,which are predominantly cytoplasmic and modify free histones,and(2) type-A,which are primarily nuclear and can be broadly classifiedinto the GNAT,MYST,and CBP/p300families.KATs were thefirst enzymes shown to modify histones.The importance of thesefindings to cancer was immediately apparent,as one of these enzymes,CBP,was identified by its ability to bind the transforming portion of the viral oncoprotein E1A(Bannister and Kouzarides,1996).It is now clear that many,if not most,of the KATs have been implicated in neoplastic transformation,and a number of viral oncoproteins are known to associate with them.There are numerous examples of recur-rent chromosomal translocations(e.g.,MLL-CBP[Wang et al., 2005]and MOZ-TIF2[Huntly et al.,2004])or coding mutations (e.g.,p300/CBP[Iyer et al.,2004;Pasqualucci et al.,2011]) involving various KATs in a broad range of solid and hematolog-ical malignancies(Figure3).Furthermore,altered expression levels of several of the KATs have also been noted in a range of cancers(Avvakumov and Coˆte´,2007;Iyer et al.,2004).In some cases,such as the leukemia-associated fusion gene MOZ-TIF2,we know a great deal about the cellular conse-quences of this translocation involving a MYST family member. MOZ-TIF2is sufficient to recapitulate an aggressive leukemia in murine models;it can confer stem cell properties and reacti-vate a self-renewal program when introduced into committed hematopoietic progenitors,and much of this oncogenic potential is dependent on its inherent and recruited KAT activity as well as its ability to bind to nucleosomes(Deguchi et al.,2003;Huntly et al.,2004).Despite these insights,the great conundrum with regards to unraveling the molecular mechanisms by which histone acetyl-transferases contribute to malignant transformation has been dissecting the contribution of altered patterns in acetylation on histone and nonhistone proteins.Although it is clear that global histone acetylation patterns are perturbed in cancers(Fraga Figure 3.Cancer Mutations Affecting Epigenetic Regulators Involved in Histone AcetylationThese tables provide somatic cancer-associated mutations identified in histone acetyltransferases and proteins that contain bromodomains(which recognize and bind acetylated histones).Several histone acetyltransferases possess chromatin-reader motifs and,thus,mutations in the proteins may alter both their catalytic activities as well as the ability of these proteins to scaffold multiprotein complexes to chromatin.Interestingly,sequencing of cancer genomes to date has not identified any recurrent somatic mutations in histone deacetylase enzymes.Abbreviations for the cancers are as follows: AML,acute myeloid leukemia;ALL,acute lymphoid leukemia;B-NHL,B-cell non-Hodgkin’s lymphoma;DLBCL,diffuse large B-cell lymphoma;and TCC, transitional cell carcinoma of the urinary bladder.Mutation types are as follows:M,missense;F,frameshift;N,nonsense;S,splice site mutation;T, translocation;and D,deletion.Cell150,July6,2012ª2012Elsevier Inc.17。
扫荡癌症基因

★ 本刊记者/蔡如鹏新年伊始,一篇发表于国际权威科学媒体《自然》的文章给全人类带来好消息:科学家一次性发现了120多个癌症基因。
自从癌症与基因的关系被证实以来,癌症基因就一直是科学家们筛淘的“金子”。
他们希望找到这些基因,进而通过调控它们,到达防治癌症的目的。
因此,每一次癌症基因的发现,都使研究人员欢欣鼓舞,觉得朝攻克癌症的终极目标又近了一步。
这一次的发现则有点出乎科学家所料,因为他们发现的不是几粒“金子”,而是一个“金矿”——上百个癌症基因。
更重要的是,这项发现是癌症基因组计划的产物,它证明科学界已经开辟出了一条“批量”发现癌症基因的路途——尽管部分科学家对此仍然存有疑虑:这些耗费巨资发现的基因,对攻克癌症究竟会有多大的帮助?它要到什么时候才会发挥作用?癌症基因集体亮相1969年,美国科学家罗伯特·许布纳和乔治·托达罗提出了一种假说,认为在所有的细胞中都含有能够导致细胞癌变的基因,这些癌症基因代代相传。
但在通常情况下它们处于被阻遏状态,只有当细胞内有关的调节机制遭到破坏的时候下,癌症基因才会“作恶”,导致癌变的发生。
现在,这一观点已经被科学界普遍接受。
在此基础上,科学家们也逐渐达成了共识:要破译癌症之谜,首先要破译基因之谜。
因为,癌症与基因是一对“亲兄弟”。
在过去的25年中,科学家已经在人类基因组的2.5万个基因中识别出了350多个与癌症有关的基因。
这些基因的发现主要是依靠扫描定位、生物鉴定等传统方法。
而这一次所用的方法不同以往,它是通过大规模的测序来发现目标基因。
负责国、中国、美国、荷兰、澳大利亚、比利时等多个国家的科学家。
他们在3月8日出版的《自然》杂志上报道说,他们对200份乳腺、胃、结肠、直肠及其他常见肿瘤样本中一类名为“激酶”的基因家族进行了研究。
激酶家族包括500多种不同的基因,它们功能的丧失是肿瘤的一个常见诱因。
HEALTH健康作为人类基因组计划向医学等实践领域的延伸,癌症基因组计划将有助于人类真正破解癌症致病的机理,其工作量与创新程度将远超过人类基因组计划扫荡癌症基因这项研究工作的英国科学家迈克·斯特拉顿来自剑桥大学著名的桑格研究所,他同时也是人类基因组计划的领导人之一。
癌症发现史

癌症发现史癌症是一种古老的疾病,考古学家在古埃及木乃伊和远古手稿中均发现了癌症存在的证据。
早在公元前2625年,古埃及伟大的医生印和阗就在莎草纸上描述了乳腺癌病例,在”治疗”项中,他只写了短短的一句:”没有治疗方法。
”1862年,埃德温·史密斯(EdwinSmith)从埃及卢克索(Luxor)的一位古董商那里买了(也有人说是偷了)一张四米多长的埃及莎草纸。
这片莎草纸状况不佳,破碎泛黄的页面上写满了潦草的古埃及文字。
这篇古文稿于1930年被翻译出来,现在被认为可能是公元前2625年古埃及伟大的医生印和阗的教诲。
莎草纸上记述了48个病例,包括了手部骨折、皮肤的多孔洞溃疡、头骨破碎,印和阗是这样描述第45个病例的:乳房上鼓起的肿块,又硬又凉,且密实如河曼果,潜伏在皮肤下蔓延——很难再找到对乳腺癌这么生动的描述了。
在”治疗”项中,他只写了短短的一句:”没有治疗方法。
”肿瘤,以及它周身蔓生的血管,让希波克拉底联想到挥脚掘沙的螃蟹“癌症(cancer)”一词的起源公元前约400年,被尊为”西方医学之父”的古希腊医师希波克拉底(Hippocrates)用”carci nos”和”carcinoma”描述肿瘤,在希腊语中,这些词语意指”螃蟹”。
肿瘤,以及它周身蔓生的血管,让希波克拉底联想到挥脚掘沙的螃蟹。
罗马医师塞尔苏斯(Celsus)随后将其翻译为”cancer”,在拉丁文中代表”螃蟹”,这也是我们今天所说的”癌症(cancer)”一词的起源。
另一名罗马医师克劳迪亚斯·盖伦(Claudius Galen)在公元前约160年用”oncos”一词描述肿瘤,在希腊语中意指”肿胀”,现代肿瘤学用语”oncology”正是脱胎于此字。
“黑色的胆汁淤积不化,遂生癌症”古希腊人眼中的癌症——黑胆汁古希腊人没有显微镜,他们从来没有见过,也想象不到存在一种叫”细胞”的实体。
为了解释所有的疾病,希波克拉底推出了一整套以液体及其体积为基础的理论,他指出,人体主要是由四种被称为”体液”的液体构成,包括血液(红)、黑胆汁(黑)、黄胆汁(黄)和黏液(白),人体在健康状态下,这四种液体处于完全的平衡中(但可能并不稳定)。
一位40年经验的癌症专家

一位40年经验的癌症专家:不要再上癌症的当!癌症专家——近藤诚日本庆应大学医院放射科医生,行医超过40年,敢于发表许多与大众切身相关却无人敢说的医疗意见,被日本民众亲切地称为“医界良心”。
出生于医生世家,毕业于庆应大学医学部,后前往美国留学,并取得博士学位。
回国后担任庆应大学医学部放射线科讲师,专攻癌症的放射线治疗,也是闻名全国的乳房保留疗法的先驱。
其功绩受到了全社会的高度评价,于2012年荣获“第60届菊池宽奖”(得奖者皆为对日本文化有莫大贡献的各界人士)。
著有《癌症别急着开刀》《患者啊,不要与癌症斗争》《癌症的放任自流疗法》等多部敲响医疗警钟的畅销著作。
1.可怕的不是癌症,而是“癌症的治疗”。
为什么一些原本很精神的人,得了癌症之后却撑不了多久呢?这都是因为他们接受了“癌症的治疗”。
只要“不治疗”癌症,患者就会保持头脑清晰的状态,直到生命的最后一刻。
只要处理方法得当,身体也能活动自如。
有许多癌症是不会痛的。
真的痛了,疼痛也是可以控制的。
如果你没有出现痛、难受、吃不下饭等症状,却在公司组织的体检中查出了癌症,那么这个“癌”就一定是“假性癌”。
光靠乳房X光片就能查出的乳腺癌也有99%是假性癌,但大多数患者仍会进行乳房切除手术,请大家多加小心。
2.真性癌发现得再早也没用。
在第一个癌症干细胞诞生的那一刻,癌症夺走宿主生命的时间就已确定下来,只不过因为发现得早,表面上的“生存时间”相应变长罢了。
所以在很多情况下,我们必须看“10年生存率”,才能判断一个患者到底有没有被“治好”。
3.手术是人工造成的重伤。
做完手术后,体力会下降,身体会很容易受感染,还有可能留下终生无法治好的后遗症,死在手术台上也是常有的事。
要是医生建议你做手术,那你最好仔细想一想,做完手术后会怎么样,想得越具体越好。
而且手术很有可能激发癌症。
医学界有一种形象的说法:“一动手术,癌细胞就会大爆发,就会暴跳如雷。
”因为手术会留下伤口,而伤口破坏了正常细胞的壁垒,血液中的癌细胞会乘虚而入,加速繁殖,最终大爆发。
山中伸弥发明IPS细胞的介绍及启示

数学统计:Pearson相 检查,发现其能够分化 Figure 5. Pluripotency of
关分析
为多种组织器官,说明 iPS Cells Derived from
其具有多能性。
MEFs
论点
证明方法
证明过程
论文对应部分
不仅是小鼠胚胎成纤 维细胞(MEFs)可被 诱导,其他分化程度
很高的体细胞也可被 诱导。
同上
将这四种因子又一起 Figure 6. 导入了小鼠尾尖成纤 Characterization of iPS 维细胞(TTFs),并进 Cells Derived from 行了与之前相同的检 Adult Mouse Tail-Tip 测,发现同样可以诱 Fibroblasts 导形成IPS细胞。
胞的多功能干细胞。 表观遗传学:染色质免 之处和差异发现ips细 iPS Cells
疫沉淀分析
胞只是类似ES细胞; Figure 4. Global Gene-
组织学:裸鼠皮下移植、同时对IPS细胞发育形 Expression Analyses by
免疫组化染色
成的畸胎瘤进行组织学 DNA Microarrays
组合在一起才能完成诱 导,其余情况均不可。
将24种因子全部加入小 Figure 1. Generation of 鼠MEF细胞中进行诱导 iPS Cells from MEF 可以得到IPS细胞,然 Cultures via 24 Factors 后将每种因子逐个去除,Figure 2. Narrowing
观察去除掉该因子后细 down the Candidate
胞能否诱导成功,最终 Factors 筛选出4个。
通过这四种转录因子的 分子生物学:RT-PCR、 研究IPS和ES在细胞形 Figure 3. Gene-
Method and the inspection kit which inspect the se
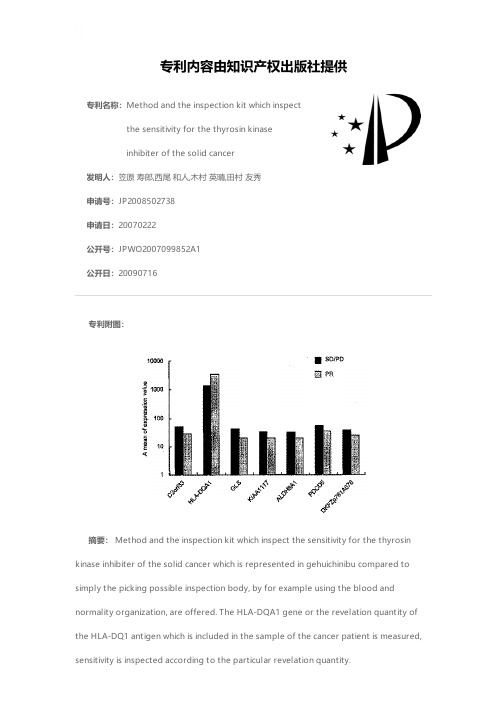
专利名称:Method and the inspection kit which inspectthe sensitivity for the thyrosin kinaseinhibiter of the solid cancer发明人:笠原 寿郎,西尾 和人,木村 英晴,田村 友秀申请号:JP2008502738申请日:20070222公开号:JPWO2007099852A1公开日:20090716专利内容由知识产权出版社提供专利附图:摘要: Method and the inspection kit which inspect the sensitivity for the thyrosin kinase inhibiter of the solid cancer which is represented in gehuichinibu compared to simply the picking possible inspection body, by for example using the blood andnormality organization, are offered. The HLA-DQA1 gene or the revelation quantity of the HLA-DQ1 antigen which is included in the sample of the cancer patient is measured,sensitivity is inspected according to the particular revelation quantity.申请人:国立大学法人金沢大学,財団法人ヒューマンサイエンス振興財団地址:石川県金沢市角間町ヌ7番地,東京都中央区日本橋小伝馬町13-4国籍:JP,JP代理人:平木 祐輔,石井 貞次,藤田 節,島村 直己更多信息请下载全文后查看。
BIOMARKER FOR PREDICTING THERAPEUTIC EFFECT OF FST

专利名称:BIOMARKER FOR PREDICTINGTHERAPEUTIC EFFECT OF FSTL1 INHIBITORIN CANCER PATIENT发明人:KUDO, Chie,工藤千恵,TOYOURA, Masayoshi,豊浦雅義,ISHIDA, Akiko,石田有希子,SHOYA, Yuji,庄屋雄二申请号:JP2017/027916申请日:20170801公开号:WO2018/025869A1公开日:20180208专利内容由知识产权出版社提供专利附图:摘要:A method for predicting the therapeutic effect of a FSTL1 inhibitor in a cancer patient, said method being characterized by comprising the steps of: (1) measuring the expression level of FSTL1 and/or DIP2A in a sample collected from the patient; (2) comparing a measurement value with a reference value; and (3) determining that the therapeutic effect is promised when the measurement value is higher than the reference value. For example, according to the present invention, a companion diagnosis for predicting the efficacy or the like of a treatment with a FSTL1 inhibitor can be performed suitably.申请人:PHARMA FOODS INTERNATIONAL CO., LTD.,株式会社ファーマフーズ地址:〒6158245 JP,〒6158245 JP国籍:JP,JP代理人:IWATANI, Ryo,岩谷龍更多信息请下载全文后查看。
ANTI-NASH COMPOSITION, FOOD COMPOSITION FOR PREVEN

专利名称:ANTI-NASH COMPOSITION, FOODCOMPOSITION FOR PREVENTING NASH,BEVERAGE COMPOSITION FOR PREVENTINGNASH, COMPOSITION FOR PREVENTINGCIRRHOSIS, AND COMPOSITION FORPREVENTING HEPATOCELLULARCARCINOMA发明人:WATANABE, Kenichi,渡辺賢一,KONISHI,Tetsuya,小西徹也,KOGA, Yusuke,古賀祐介申请号:JP2018/003958申请日:20180206公开号:WO2018/147260A1公开日:20180816专利内容由知识产权出版社提供专利附图:摘要:This anti-NASH composition, this food composition for preventing NASH, and this beverage composition for preventing NASH include, as an active ingredient, a Basidiomycetes-X FERM BP-10011 dry powder or an extract composition thereof. In addition, a composition for preventing cirrhosis and a composition for preventing hepatocellular carcinoma include, as an active ingredient, a Basidiomycetes-X FERM BP-10011 dry powder or an extract composition thereof, and prevent metastasis of cirrhosis and hepatocellular carcinoma from NASH.申请人:MYCOLOGY TECHNO.CORP.,マイコロジーテクノ株式会社地址:4-8, Yamakido 8-chome, Higashi-ku, Niigata-shi, Niigata 9500871 JP,〒9500871新潟県新潟市東区山木戸八丁目4番8号 Niigata JP国籍:JP,JP代理人:KURIHARA, Hiroyuki et al.,栗原浩之更多信息请下载全文后查看。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Deep Sequencing in Cancer ResearchKenichi Yoshida,Masashi Sanada and Seishi Ogawa *Cancer Genomics Project,Graduate School of Medicine,The University of Tokyo,Tokyo,Japan *For reprints and all correspondence:Seishi Ogawa.E-mail:sogawa-tky@umin.ac.jpReceived August 26,2012;accepted November 4,2012Cancer is caused by alterations in the cellular genome including single-nucleotide variations,small insertions and deletions (indels),copy number changes and other structural variations and,as such,their detection in a comprehensive manner is of critical importance for fully understanding cancer pathogenesis,improvement of diagnosis as well as the development of novel therapeutics.In this point of views,the recent development of massively parallel (or ‘next-generation’)sequencing technologies has provided an unprecedented possibility to ac-complish this need by enabling single-nucleotide resolution analysis of the entire genome of cancer cells as well as more targeted analysis of coding sequencing or transcriptomes.Through international co-operations,a wide variety of cancer cell types have now been analyzed using these technologies to help unmask their pathogenesis.In this review,we briefly overview the recent advances in cancer research obtained through the massive effort of sequencing cancer genomes.Key words:massively parallel sequencing –cancer genome –somatic mutationINTRODUCTIONCancer is caused by accumulation of genomic abnormalities (1).Therefore,comprehensive detection of various types of genetic changes,such as point mutations,copy number alterations and structural variations,are critically important for the understanding cancer biology,the improvement of diagnosis as well as the development of targeted therapies.Cancer genomes are highly complex,which are composed of as many as 3billion bases,including 24000–25000of protein-coding genes.The initial clue to accommodate this complexity of cancer genomes was obtained by the construc-tion of the first draft sequence of the human genome in 2001through the international collaborative effort under the human genome project (2).Since then,the sequence has been used as the reference human genome,based on which researches have been analyzing somatic mutations in cancers.However,the real breakthrough to facilitate the de-tection of cancer-specific gene mutations/alterations has been brought about by the recent development of massively parallel sequencing technologies;the 13years requiredto read an initial human genome has been dramatically reduced to 2weeks with typical next-generation sequen-cing.This revolution of sequencing technologies envisages a new era of cancer genetics and biology.In this review,we overview the second-generation sequencing technologies and their application to cancer research.ANALYSIS OF CANCER GENOMES USING SECOND-GENERATION SEQUENCING TECHNOLOGIESA typical second-generation (also known as ‘next-generation’)sequencer goes through billions of fragmented DNAs (reads)simultaneously in a highly parallel way (mas-sively parallel sequencing).Inevitably high sequencing errors in reading numerous bases are compensated by reading multiple (.30–40times)DNA fragments for an ac-curate determination of somatic mutations and allelic status of polymorphisms in diploid sequences.For example,typic-ally 90Gb of sequence or 30-fold coverage are obtained#The Author 2012.Published by Oxford University Press.All rights reserved.For Permissions,please email:journals.permissions@doi:10.1093/jjco/hys206Advance Access Publication 5December2012at University of Melbourne Library on September 25, 2014/Downloaded fromto call99%of SNP alleles(3).The need of multiple reads is also realized by the fact that the mutations under interest may exist only in low allele frequencies in many situations; primary cancer samples are more or less‘contaminated’with normal tissues and there also exist multiple tumor subclones with different genetic alterations within a tumor,comprising intra-tumor heterogeneity(4–6).It may be necessary to deeply sequence to capture such low-frequency alleles de-pending on the apparent frequencies of the relevant alleles. To further compound matters,pathogenesis of cancer is heterogeneous in terms of the spectrum of gene mutations even within the same histology type.Thus,sequencing mul-tiple tumor samples may be necessary to understand the full spectrum of causative mutations depending on the extent of the heterogeneity,except for rare cases where the tumor is highly homogeneous and therefore only a few samples were sufficient to capture the relevant mutations.In fact,several ongoing projects are analyzing large numbers of cancer spe-cimens;in the Cancer Genome Atlas(TCGA)(7)promoted by NIH/NCI in the USA and the International Cancer Genome Consortium(ICGC)participated in by institutes from multiple nations(8),more than500specimens for each of the major cancer types will be subjected to integrated genome,epigenome,transcriptome studies(9–14). WHOLE-GENOME SEQUENCINGIn2008,thefirst sequencing of an entire cancer genome was reported,providing a comprehensive registry of gene mutations in a case with acute myeloid leukemia(AML) with a normal karyotype.In whole-genome sequencing,fragmented genomic DNA is directly subjected to massively parallel sequencing after minimum processing with several cycles of PCR amplification.Although simple in manipula-tion,this approach provides least biased,full-range informa-tion about genetic alterations at a single-nucleotide level (Fig.1).Major caveats include the cost and time required for the analysis,compared with sequencing with lower complex-ity.It also consumes much more computer resources. Because of its uniform coverage across all the genomic regions except for GC-rich sequences,whole-genome se-quencing captured mutations that occur in both coding and non-coding regions,including promoters,enhancer,introns and non-coding RNAs as well as intergenic sequences.Not only detecting single-nucleotide substitutions,whole-genome sequencing can also identify a wide variety of structural ab-normalities,such as chromosomal translocations,deletions and insertions,as well as copy number alterations,in a com-prehensive manner.Especially,currently no other methods are available for detecting chromosomal translocations sys-tematically,which would be underscored by the fact that a number of fusion genes caused by chromosomal transloca-tions play a critical role in the cancer pathogenesis and are also exploited for contriving novel diagnostics and therapeu-tics(15,16).Moreover,whole-genome sequencing can reveal a more complex aspect of cancer genomes,in which tens to hundreds of genomic rearrangements involved localized genomic regions(‘chromothripsis’),which were thought to be acquired in an apparently one-off cellular crisis and reported to occur in2–3%of all cancers and 25%of bone cancers(Fig.2)(17).Finally,whole-genome sequencing is capable of detecting non-human sequencing derived from microbial genomebyFigure1.Various types of genetic alterations that can be detected by whole-genome sequencing.Sequenced fragments(reads)are depicted as bars andpaired-end reads connected with a dotted line are mapped to different genomic positions in the case of translocation.Copy number alterations can be detectedby change in the number of sequence reads(depth)compared with a normal control.at University of Melbourne Library on September 25, 2014/Downloaded fromanalyzing sequences that are not mapped to human reference genome.Sung et al.performed whole-genome sequencing of 76HBV-positive hepatocellular carcinomas and detected HBV integration in 76.4%of tumor samples.They also revealed that HBV integrations in TERT ,MLL4and CCNE1genes accounted for 40%of cases with HBV integrations (18).As the cost of sequencing continues to reduce,whole-genome sequencing is becoming a standard method of cancer genome sequencing feasible for many researchers.TARGETED/WHOLE-EXOME SEQUENCINGConsidering the current cost and throughput of sequencing,alternative to whole-genome sequencing would be focusing on particular sequences under interest,for example,entire coding sequences that account for ,1.3%of the total genomes,which are now widely used for cancer genome se-quencing (targeted sequencing).In whole-exome sequences,entire protein-coding sequences encompassing from 50to 62Mb are captured using liquid–solid phase hybridization to chemically synthesized nucleotides or ‘baits’(Fig.3),which were subjected to massively parallel sequencing.By limiting the targets to approximately one-hundredth of the entire genome,much higher sequence coverage ( 100times)can be achieved with considerably less sequence data compared with whole-genome sequencing,which far outperforms capillary sequencing-based approach employed in exome sequencing of glioblastoma (19).Potential drawbacks in whole-exome sequencing are add-itional costs and procedures required for target capture andcompromised sensitivity due to less uniform coverage and inability to detect fusion genes and other structural varia-tions.Nevertheless,whole-exome sequencing provides a rea-sonable alternative to whole-genome sequencing,in terms of a high throughput of analyzing multiple samples at higher coverage and lower costs and the higher sensitivity of detect-ing mutations with lower allele frequencies,which may exist only in minor subclones or be associated with lower tumor burden,although it is challenging to detect somatic muta-tions in cases where germline control sample is contami-nated with tumor tissues.In fact,whole-exome sequencing has been successfully applied to identify a number of novel targets of recurrent mutations (10,20–24).TRANSCRIPTOME SEQUENCINGOne of the major shortcomings in whole-exome sequencing is its inability to detect gene fusions.The deficit could be complemented in part by transcriptome sequencing (RNA sequencing,RNA-seq)at an acceptable cost (Fig.4).In fact,transcriptome sequencing has been shown to provide a powerful method to detect biologically relevant fusion genes in that the detected fusion genes are actually expressed in tumor cells.The exact sequences of the expressed fusion transcripts can be directly determined.Steidl et al.performed RNA-seq of Hodgkin lymphoma cell lines and identified gene fusion involving MHC class II transactivator CIITA (25),which were also found in primary mediastinal B-cell lymphoma (38%)and Hodgkin lymphoma samples (15%).Several algorithms are available for detecting fusiongenesFigure 2.Schematic overview of chromothripsis and a circos plot of a chromothripsis-positive cancer.(A)Schematic overview of chromotripsis.(B)Chromothripsis detected in a case of clear cell renal carcinoma by whole-genome sequencing.Chromosomes (outside of the circular plot)and chromosom-al rearrangements are shown as arcs connecting the two relevant genomic regions in the middle.Chromothripsis rearrangements are indicated by green arrows.112Cancer research and deep sequencingat University of Melbourne Library on September 25, 2014/Downloaded frombased on transcriptome sequencing,many of which are no-torious for high false-positive rates even for recurrently detected candidates,which may necessitate intensive valid-ation tasks using RT–PCRs.RNA-seq can be used to detect somatic mutations and some important discovery of recurrent mutations in cancer has been made by RNA-seq,such as FOXL2mutations in ovarian granulosa cell tumors (26).Although biologically relevant mutations tend to be enriched expressed sequences,coverage of sequencing could be highly biased depending on expression levels of different targets,leading to loss of power for detecting some mutations.This is in contrast to whole-genome/exome sequencing,with which relatively un-biased detection is expected.Difficulty in obtaining appro-priate normal control tissues is another significant problem with transcriptome-based detection of mutations.RNA-seq also allows analysis of gene expression profiles in highersensitivity.Novel transcripts or alternative splicing forms could be detected also,although computational analysis could be a challenging task.However,the higher representa-tion of 30sequences could prevents unbiased estimation of relative expression levels,especially estimation of large tran-scripts,in which significant underrepresentation of the 50sequences occurs.DEEP SEQUENCING OF SELECTED TARGETSIntratumoral heterogeneity seems to be a common feature of cancers,providing a major source of tumor recurrence or re-sistance to anti-cancer therapies.It could be best evaluated by deep sequencing in combination with whole-genome/exome sequencing,where accurate allele frequencies of a set of mutations detected by genome/exome sequencingareFigure 3.Targeted sequencing using in-solution captureapproach.Figure 4.FUS -DDIT3fusion gene of liposarcoma detected by transcriptome sequencing.Jpn J Clin Oncol 2013;43(2)113at University of Melbourne Library on September 25, 2014/Downloaded fromdetermined by reading at higher depth(typically.1000 or10000).Target DNA was amplified by individual PCRs or captured by high-throughput hybridization to the target baits.As low as1%of minor alleles could be sensitively detected by target deep sequencing depending on experimen-tal conditions and sequencing platforms,although back-ground errors inherent to sequencing chemistries would prevent further detection of even lower allele frequencies. Saha et al.performed deep sequencing(median20000 times)of somatic mutations in triple-negative breast cancers detected by whole-exome sequencing,in which existence of subclones with different mutant allele frequencies were clearly demonstrated in most of cases(5).Walter et al.also showed the presence of subpopulation in secondary AML cases derived from myelodysplastic syndromes by whole-genome sequencing followed by target deep resequeing (average640times)of304–872candidates of somatic muta-tions using solid-phase target capture(6).Another application of target deep sequencing is mutation screening involving a large number of genes and tumor samples.Actually,it becomes more and more unrealistic in detecting mutations of a number of large genes among hun-dreds of specimens using Sanger sequencing and plausible application of high-throughput sequencing.Deep sequencing also enabled a sensitive detection of minor mutant alleles with low frequencies together with an accurate determination of allele frequencies at the same time(10,20,27). SUMMARY AND FUTURE DIRECTIONS Considering the ever-decreasing cost for sequencing,major mutational targets will be identified for common cancer types within a few years.However,many issues should be remained to be unanswered.For many newly identified mutations,their biological/clinical significance as well as their molecular mechanics in cancer development is unknown.There is an urgent need to achieving the clinical application of these technologies for diagnosis and treatment stratification,eventually leading to cure for cancer,although special attention should be paid before clinical use.For example,validation of the candidate mutations using alterna-tive methods such as the conventional Sanger sequencing is indispensable at the current moment considering a relatively high error rate of second-generation sequencing.While understanding of the intratumoral heterogeneity is one of the critical issues in carcinogenesis,especially with regard to re-currence or resistance to anti-cancer therapies,higher reso-lution/throughput sequencing technologies,ideally of single-cell genomes,will be required to approach this issue. Promising news is the recent announcement of the coming newer generation sequencing platforms,including Ion tor-rent’s and Nanopore technologies(28,29).Roles of epigenet-ic abnormalities together with their interactions with gene mutations need to be clarified in further details,for which another technological breakthrough should be warranted.FundingThis work was supported by Grant-in-Aids from the Ministry of Health,Labor and Welfare of Japan and from the Ministry of Education,Culture,Sports,Science and Technology,and the Japan Society for the Promotion of Science(JSPS)through the‘Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program),’initiated by the Council for Science and Technology Policy(CSTP)and also by‘project for develop-ment of innovative research on cancer therapies(p-direct)’. Conflict of interest statementNone declared.References1.Stratton MR,Campbell PJ,Futreal PA.The cancer genome.Nature2009;458:719–24.nder ES,Linton LM,Birren B,et al.Initial sequencing and analysisof the human genome.Nature2001;409:860–921.3.Li H,Ruan J,Durbin R.Mapping short DNA sequencing reads andcalling variants using mapping quality scores.Genome Res2008;18:1851–8.4.Gerlinger M,Rowan AJ,Horswell S,et al.Intratumor heterogeneity andbranched evolution revealed by multiregion sequencing.N Engl J Med 2012;366:883–92.5.Shah SP,Roth A,Goya R,et al.The clonal and mutational evolutionspectrum of primary triple-negative breast cancers.Nature2012;486:395–9.6.Walter MJ,Shen D,Ding L,et al.Clonal architecture of secondaryacute myeloid leukemia.N Engl J Med2012;366:1090–8.7./.8./.9.Integrated genomic analyses of ovarian carcinoma.Nature2011;474:609–15.10.Papaemmanuil E,Cazzola M,Boultwood J,et al.Somatic SF3B1mutation in myelodysplasia with ring sideroblasts.N Engl J Med 2011;365:1384–95.11.Wang L,Lawrence MS,Wan Y,et al.SF3B1and other novel cancergenes in chronic lymphocytic leukemia.N Engl J Med2011;365:2497–506.prehensive molecular characterization of human colon and rectalcancer.Nature2012;487:330–7.13.Fujimoto A,Totoki Y,Abe T,et al.Whole-genome sequencing of livercancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators.Nat Genet2012;44:760–4.14.Jones DT,Jager N,Kool M,et al.Dissecting the genomic complexityunderlying medulloblastoma.Nature2012;488:100–5.15.Tomlins SA,Rhodes DR,Perner S,et al.Recurrent fusion of TMPRSS2and ETS transcription factor genes in prostate cancer.Science 2005;310:644–8.16.Soda M,Choi YL,Enomoto M,et al.Identification of the transformingEML4-ALK fusion gene in non-small-cell lung cancer.Nature2007;448:561–6.17.Stephens PJ,Greenman CD,Fu B,et al.Massive genomicrearrangement acquired in a single catastrophic event during cancer development.Cell2011;144:27–40.18.Sung WK,Zheng H,Li S,et al.Genome-wide survey of recurrent HBVintegration in hepatocellular carcinoma.Nat Genet2012;44:765–9. 19.Parsons DW,Jones S,Zhang X,et al.An integrated genomic analysisof human glioblastoma multiforme.Science2008;321:1807–12.20.Yoshida K,Sanada M,Shiraishi Y,et al.Frequent pathway mutations ofsplicing machinery in myelodysplasia.Nature2011;478:64–9.21.Tiacci E,Trifonov V,Schiavoni G,et al.BRAF mutations in hairy-cellleukemia.N Engl J Med2011;364:2305–15.114Cancer research and deep sequencingat University of Melbourne Library on September 25, 2014/Downloaded from22.Pasqualucci L,Dominguez-Sola D,Chiarenza A,et al.Inactivatingmutations of acetyltransferase genes in B-cell lymphoma.Nature 2011;471:189–95.23.Agrawal N,Frederick MJ,Pickering CR,et al.Exome sequencing ofhead and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1.Science2011;333:1154–7.24.Stransky N,Egloff AM,Tward AD,et al.The mutational landscape ofhead and neck squamous cell carcinoma.Science2011;333:1157–60. 25.Steidl C,Shah SP,Woolcock BW,et al.MHC class II transactivatorCIITA is a recurrent gene fusion partner in lymphoid cancers.Nature 2011;471:377–81.26.Shah SP,Kobel M,Senz J,et al.Mutation of FOXL2in granulosa-celltumors of the ovary.N Engl J Med2009;360:2719–29.27.Puente XS,Pinyol M,Quesada V,et al.Whole-genome sequencingidentifies recurrent mutations in chronic lymphocytic leukaemia.Nature2011;475:101–5.28.Rothberg JM,Hinz W,Rearick TM,et al.An integrated semiconductordevice enabling non-optical genome sequencing.Nature2011;475:348–52.29.Branton D,Deamer DW,Marziali A,et al.The potentialand challenges of nanopore sequencing.Nat Biotechnol2008;26:1146–53.Jpn J Clin Oncol2013;43(2)115at University of Melbourne Library on September 25, 2014/Downloaded from。