测序峰图
DNA峰图

第一类突变:通过测序峰图的学习,我们找到第一次测序中 (ACGCC)与第二次测序突变位点(G→A)(ACACC)相对应的峰图
峰图比较说明,在第一次测序中该位点正常,而在第二次测序中显示为 位点突变(G→A)。但经过进一步分析密码子顺序,此处编码氨基酸的密 码子应为ACG,编码苏氨酸。而在突变成ACA后,对应编码的氨基酸并没 有改变,因此此处位点的突变没有影响。
Lab Meeting
彭军
1、pET-22b-Nek7-His6载体构建
• Nek7 PCR→胶回收→LIC连接→挑菌摇菌→菌液 PCR→质粒小提→测序 • 测序结果与NCBI查得序列100%匹配。
2.pCMV-Nek7-His6-3×flag载体构建
• ……→小提质粒后测序,DNAMAN比对,比对结果出入有两类。 一类是在测序精度范围之内的一个位点(基因的597位)突变 (G→A),另一类是在测序精度范围之外的位点(基因的878位) 的缺失。
• 第二类缺失:由于第二类缺失发生在测序精度范围之外,对应的 峰图也不一定精确,我们也去下面测序公司询问了公司的人员, 他们说测序精度只能保证800以内,这个地方可能是没有缺失的。 因此我们直接将这个质粒转染进细胞看蛋白大小是否与理论一致。
• 峰图学习: 根据此次克隆的问题,在测序问题上,当用DNAMAN比对完全正确 时,可以不用去分析峰图。但是当测序比对出现问题时,需要回到 峰图去分析机器读峰的准确性以及有无杂峰干扰等问题。因为测得 的序列都是通过峰图转换成序列的。 测序公司发送的文件中会包含一个峰图ab1文件,用Chromas软件 打开可以观测分析峰图。
3.转染细胞看表达
• 经过PEI转染进293T细胞,并以转染Pcmv-3×flag空载作为对照。 第二天观察,发现实验组细胞并没有明显的死亡。随后收细胞用 200ul裂解液裂解后做WB,得到以下结果:
TALEN和CAS9技术移码敲除测序峰图分析方法说明-仅供参考

1.为什么TALEN和CAS9技术移码敲除项目需通过测序判定基因型?TALEN和CAS9技术是通过特异识别和核酸酶切割,使DNA双链断开,机体启动DNA损伤修复机制,在非同源末端连接修复过程中,碱基的随机增减造成目标基因功能缺失。
这一修复过程通常会产生1-30个左右的碱基缺失,PCR后凝胶电泳无法将碱基缺失链和wt链区分开,所以只能通过测序判断。
2.如何判断TALEN和CAS9技术移码敲除项目的基因型?a. 野生型或基因敲除的纯合子基因型的判定:1)如下图所示,只有单峰的为野生型或基因敲除的纯合子。
2)将只有单峰的序列文件与wt序列比对,可判定野生型或基因敲除的纯合子基因型(网站或生物软件比对均可).如下图的对比结果为野生型:如下图的比对结果为缺失一个C的纯合子b.杂合子基因型的判定:1).如下图所示,测序图谱中出现叠峰的为杂合子若将叠峰的序列文件直接与wt序列比对,叠峰后的序列会对应不上(因为叠峰的序列只显示了信号强一点的那个碱基,实际上每个叠峰对应两个碱基),如下图所示:所以不能通过直接比对来分析,需通过峰图分析,在峰图中峰的颜色与碱基对应关系如下:A:绿色T:红色C:蓝色G:黑色2)将wt的峰图与杂合子的峰图用软件同时打开,从出现叠峰的位置开始分析(杂合子的一条链是wt链,一条链是缺失链):上图第一个峰图是野生型的序列,与第二个峰图叠峰开始对应的序列为:Gttaact ccgagcagcaaagaaatgatgtccc上图第二个峰图是杂合子的测序峰图,从叠峰位置开始的序列为:Gttaact ccgagcagcaaagaaatgatgtccc(wt链)Ccgagcagcaaagaaatgatgtcccaagcctt(缺失链)由此可判定为该杂合子的基因型为缺失gttaact(-7)。
套峰

套峰套峰:即主峰下面套有低于或与主峰高度一致的干扰峰,如果干扰峰信号较强,则会影响主峰的正确读值。
引起套峰的原因包括以下一种或多种原因的组合:1. 测序模板非单一:a. PCR产物不纯:PCR模板中含有点突变,如下图:峰图说明:峰图其他位点碱基峰形独立且单一,只有个别位点有两个信号峰套在一起,如箭头所示。
常见原因为PCR的模板DNA中含有突变或者SNP多态性的等位基因。
序列插入缺失突变,如下图:峰图说明:起始峰图正常,但从某一位置开始出现套峰,套峰序列与主峰序列基本相同,但出现碱基的移位;常见原因为PCR的模板DNA中含有插入后者缺失突变的等位基因。
PCR扩增非特异,PCR产物不纯,如下图:峰图说明:起始峰图即为套峰,可以看到多个测序终止峰型,如箭头所示。
b.质粒不纯或者非单一克隆:质粒包含非单一克隆质粒,如下图:峰图说明:峰图起始正常,但随后出现套峰,套峰较高时无法辨认主峰。
2. 测序引物不纯或者污染:可能来自于引物降解,如下图:峰图说明:底峰读值与下一个或上一个主峰一致,常见原因为测序引物的降解,部分引物分子丢失了3’端核苷酸。
PCR产物中含有引物二聚体,如下图:峰图说明:起始峰图即为套峰或者底峰,一般至150bp以内会有明显“A尾”终止峰形(如箭头所示),随后峰图正常。
这是由于PCR产物中的引物二聚体未被去除,与测序引物结合产生信号所至。
PCR引物残留,如下图:峰图说明:峰图从起始到结束均有套峰。
形成原因为PCR引物未被去除,在测序反应中被作为测序引物与模板结合并延伸。
引物与模板结合非单一:在测序反应的退火温度与模板和测序引物的比例条件下,引物与模板有多个结合位点。
峰图说明:峰图从起始到结束均有套(底)峰。
解决套峰的方法:1.针对测序模板不纯和非单一克隆,解决的方式为尽量得到单一的纯化的测序模板:a)针对PCR产物:1)优化PCR条件或者重新设计PCR引物,提高引物扩增的特异性;2)如果PCR产物条带不单一,可采用切胶回收的方式纯化后测序;3)如果PCR产物条带单一,但测序结果仍有套峰,可将PCR产物克隆后测序;4)存在等位基因多态性的模板的扩增产物,如果套峰影响碱基序列读取,也可以考虑将PCR产物克隆后选取多个克隆分别测序;5)设计序列特异性的测序引物,以期得到特异性的测序结果。
一代测序结果峰图的基本知识
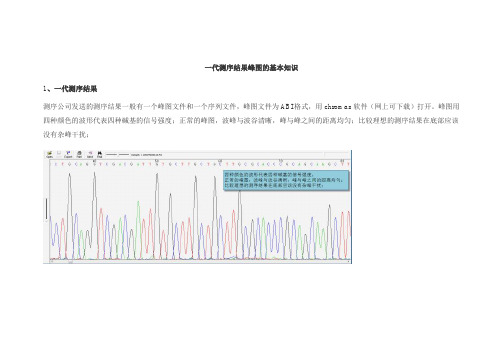
一代测序结果峰图的基本知识
1、一代测序结果
测序公司发送的测序结果一般有一个峰图文件和一个序列文件,峰图文件为ABI格式,用chromas软件(网上可下载)打开。
峰图用四种颜色的波形代表四种碱基的信号强度;正常的峰图,波峰与波谷清晰,峰与峰之间的距离均匀;比较理想的测序结果在底部应该没有杂峰干扰;
2、起始序列说明
1)测序结果的前几十bp不能用(下图中黄色区域标出的区域),在拼接序列或序列比对时需要先去除这部分不太可靠的数据,以免影响分析结果;
2)在使用PCR引物测序时由于PCR引物上的碱基是不带染料的,所以检测不到信号,在测序结果中也找不到引物的序列,如连接到载体使用载体上的通用引物测序则可以找到PCR引物;
3、有效读长
测序公司给出的峰图在1000~1200bp之间,但超过800bp后的各种波之间有重叠,波峰有钝化现象,碱基与碱基间的距离有时突然加大(此图中的812bp~813bp),这些现象都会影响结果的准确性,所以只能做为参考.所以说有效片段为800bp左右.
4、SNP位点
SNP位点为纯合时成单峰,若主杂合子时,会形成套峰现象;测序公司给的结果序列文件通常只读主峰——即序列文件不会报告SNP 杂合,需要自己打开峰图文件判读。
一代测序结果峰图的基本知识

一代测序结果峰图的基本知识1、一代测序结果
测序公司发送的测序结果一般有一个峰图文件和一个序列文件,峰图文件为ABI格式,用chromas软件(网上可下载)打开。
峰图用四种颜色的波形代表四种碱基的信号强度;正常的峰图,波峰与波谷清晰,峰与峰之间的距离均匀;比较理想的测序结果在底部应该没有杂峰干扰;
.
2、起始序列说明
1)测序结果的前几十bp不能用(下图中黄色区域标出的区域),在拼接序列或序列比对时需要先去除这部分不太可靠的数据,以免影响分析结果;
2)在使用PCR引物测序时由于PCR引物上的碱基是不带染料的,所以检测不到信号,在测序结果中也找不到引物的序列,如连接到载体使用载体上的通用引物测序则可以找到PCR引物;
.
3、有效读长
测序公司给出的峰图在
1000~1200bp之间,但超过800bp后的各种波之间有重叠,波峰有钝化现象,碱基与碱基间的距离有时突然
加大(此图中的812bp~813bp),这些现象都会影响结果的准确性,所以只能做为参考.所以说有效片段为800bp左右.
.
4、SNP位点
SNP位点为纯合时成单峰,若主杂合子时,会形成套峰现象;测序公司给的结果序列文件通常只读主峰——即序列文件不会报告SNP 杂合,需要自己打开峰图文件判读。
.。
测序常见峰图原因说明及建议解决方案

客服多安排了测序反应
如果由于公司原因导致多安排了 反应,公司将采用相应的措施。
可能是引物与模板结合有问题。 样品用完
建议核实引物是否适合模板测 序,可以考虑更换测序引物
建议客户重新提供测序样品或者 引物。
菌的活性较低或者质粒拷贝数较低 公司安排第二次摇菌或者单管摇
等样品的问题;也有可能摇菌过程 菌;您可以根据实验情况,决定
测序完成,插入后双峰,样品 非单克隆。
模板中含有两个或两个以上的相同 载体,但是插入片段不同。
重新转化后挑取单克隆测序。
菌液、质 粒、PCR 已纯化、
测序完成,Poly(A/T/C/G)结 构后出现双峰/乱峰/衰减/中断 。
您的目的序列中有连续的A/T/C/G 。
建议更换反向引物测序。
PCR未纯 化、其他 测序完成,测序结果双峰,可
建议解决的方案
实验取消,客户样品/引物异 可能PCR产物、质粒或者引物降解 建议客户重新提供模板或者引物
常,无法实验。
、质粒抽提异常。
。
实验取消,客户提供信息不 全,无法实验。
客户提供的样品信息不完整,例如 抗性、引物等信息不确定。
建议客户准确完整填写测序订 单;如填写过程中有疑问,可以 及时与我们客服联系。
建议1:将测序样品用高级试剂 盒调整;得出较好的结果,再设 计引物继续实验;2:客户提供 目的片段,我们分析是否可以继 续设计引物;3:单向测通。
实验取消,该反应多余。
菌液、质 粒、PCR 实验取消,试做结果差。 已纯化、 PCR未纯 实验取消,由于样品用完,等 化、其他 待客户重新提供样品。
类型 实验进行中,第一次摇菌不成 功,目前正在进行第二次摇菌 。
实验停止,测序结果差。
一代测序峰图(sanger)颜色标记 全称 三字 单字简写 氨基酸及对应密码子表(DNA)总表

TTTPhe F Phenylalanine TCT Ser S Serine TAT Tyr T Tyrosine TGT Cys C TTC Phe F Phenylalanine TCC Ser S SerineTAC Tyr T Tyrosine TGC Cys C TTA Leu L Leucine TCA Ser S SerineTAA stop TGA stop TTG Leu L Leucine TCG Ser S SerineTAG stop TGG Trp W CTT Leu L Leucine CCT Pro P ProlineCAT His H Histidine CGT Arg R CTC Leu L Leucine CCC Pro P ProlineCAC His H Histidine CGC Arg R CTA Leu L Leucine CCA Pro P ProlineCAA Gln Q GlTtamine CGA Arg R CTG Leu L Leucine CCG Pro P Proline CAG Gln Q GlTtamineCGG Arg R ATT Ile I Isoleucine ACT Thr T Threonine AAT Asn N AsparagineAGT Ser S ATC Ile I Isoleucine ACC Thr T Threonine AAC Asn N AsparagineAGC Ser S ATA Ile I Isoleucine ACA Thr T Threonine AAA Lys K LysineAGA Arg R ATG Met M MethionineACG Thr T Threonine AAG Lys K Lysine AGT Arg R GTT Val V ValineGCT Ala A Alanine GAT Asp D Aspartic acid GGT Gly G GTC Val V ValineGCC Ala A Alanine GAC Asp D Aspartic acid GGC Gly G GTA Val V ValineGCA Ala A Alanine GAA GlT E Glutamic acid GGA Gly G GTG Val V ValineGCG Ala A Alanine GAG GlT E Glutamic acid GGG Gly G R:A/G W:A/TV:A/G/C B:G/C/T Y:C/T K:G/T D:A/G/T M:A/C S:G/C H:A/C/T N:A/G/C/TA G TC A G T C作者:刘洪洲CysteineT Cysteine C A Trptophan G Arginine T Arginine C Arginine A Arginine G Serine T Serine C Arginine A Arginine G Glycine T Glycine C Glycine A Glycine GG。
测序峰图

峰的高低说明了信号的强弱,宽窄表示的是分离效果,可以对比板胶电泳的亮度和条带的宽窄。
重叠则说明了样品中有长度相同但是序列不同的片段。
Q-1.为什么提供新鲜的菌液?如何提供新鲜的菌液?返回顶端A-1.首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度。
如果您提供新鲜菌液,用封口膜封口以免泄露;也可以将培养好的4~5ml菌液沉淀下来,倒去上清液以方便邮寄。
同时邮寄时最好用盒子以免邮寄过程中压破。
Q-2.DNA测序样品用什么溶液溶解比较好?返回顶端A-2.溶解DNA测序样品时,用灭菌蒸馏水溶解最好。
DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。
如果DNA用缓冲液溶解后,在进行测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。
有很多客户在溶解DNA测序样品时使用TE Buffer。
的确,TE Buffer能增加DNA样品保存期间的稳定性,并且TE Buffer对DNA测序反应的影响也较小,但根据我们的经验,我们还是推荐使用灭菌蒸馏水来溶解DNA测序样品。
Q-3.提供DNA测序样品时,提供何种形态的比较好?返回顶端A-3.我们推荐客户提供菌体,由我们来提取质粒,这样DNA样品比较稳定。
如果您可以提供DNA样品,我们也很欢迎,但一定要注意样品纯度和数量。
如果提供的DNA量不够,我们就需要对质粒进行转化,此时需收取转化费。
有些质粒提取法提取的DNA质量很好,象TaKaRa、Qiagen、Promega的质粒制备试剂盒等。
提供的测序样品为PCR产物时,特别需要注意DNA的纯度和数量。
PCR产物必须进行切胶回收,否则无法得到良好的测序效果。
有关DNA测序样品的详细情况请严格参照“测序样品的提供”部分的说明。
Q-4.提供的测序样品为菌体时,以什么形态提供为好?返回顶端A-4.一般,菌体的形态有:平板培养菌、穿刺培养菌,甘油保存菌或新鲜菌液等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
峰的高低说明了信号的强弱,宽窄表示的是分离效果,可以对比板胶电泳的亮度和条带的宽窄。
重叠则说明了样品中有长度相同但是序列不同的片段。
Q-1.为什么提供新鲜的菌液?如何提供新鲜的菌液?返回顶端A-1.首先,新鲜的菌液易于培养,可以获得更多的DNA,同时最大限度地保证菌种的纯度。
如果您提供新鲜菌液,用封口膜封口以免泄露;也可以将培养好的4~5ml菌液沉淀下来,倒去上清液以方便邮寄。
同时邮寄时最好用盒子以免邮寄过程中压破。
Q-2.DNA测序样品用什么溶液溶解比较好?返回顶端A-2.溶解DNA测序样品时,用灭菌蒸馏水溶解最好。
DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件。
如果DNA用缓冲液溶解后,在进行测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。
有很多客户在溶解DNA测序样品时使用TE Buffer。
的确,TE Buffer能增加DNA样品保存期间的稳定性,并且TE Buffer对DNA测序反应的影响也较小,但根据我们的经验,我们还是推荐使用灭菌蒸馏水来溶解DNA测序样品。
Q-3.提供DNA测序样品时,提供何种形态的比较好?返回顶端A-3.我们推荐客户提供菌体,由我们来提取质粒,这样DNA样品比较稳定。
如果您可以提供DNA样品,我们也很欢迎,但一定要注意样品纯度和数量。
如果提供的DNA量不够,我们就需要对质粒进行转化,此时需收取转化费。
有些质粒提取法提取的DNA质量很好,象TaKaRa、Qiagen、Promega的质粒制备试剂盒等。
提供的测序样品为PCR产物时,特别需要注意DNA的纯度和数量。
PCR产物必须进行切胶回收,否则无法得到良好的测序效果。
有关DNA测序样品的详细情况请严格参照“测序样品的提供”部分的说明。
Q-4.提供的测序样品为菌体时,以什么形态提供为好?返回顶端A-4.一般,菌体的形态有:平板培养菌、穿刺培养菌,甘油保存菌或新鲜菌液等。
我们提倡寄送穿刺培养菌或新鲜菌液。
平板培养菌运送特别不方便,我们收到的一些平板培养菌的培养皿在运送过程中常常已经破碎,面目全非,需要用户重新寄样。
这样既误时间,又浪费客户的样品。
一旦是客户非常重要的样品时,其后果更不可设想。
而甘油保存菌则容易污染。
制作穿刺菌时,可在1.5 ml的Tube管中加入琼脂培养基,把菌体用牙签穿刺于琼脂培养基(固体)中,37℃培养一个晚上后便可使用。
穿刺培养菌在4℃下可保存数个月,并且不容易污染,便于运送。
Q-5.PCR产物直接测序有什么要求?返回顶端A-5.(1) 扩增产物必须特异性扩增,条带单一。
如果扩增产物中存在非特异性扩增产物,一般难以得到好的测序结果。
(2) 必须进行胶回收纯化。
(3) DNA纯化在1.6~2.0之间,浓度50ng/µl以上。
Q-6.为什么PCR产物直接测序必须进行Agarose胶纯化?返回顶端A-6.如果不进行胶纯化而直接用试剂盒回收,经常会导致测序出现双峰甚至乱峰。
这主要是非特异性扩增产物或者原来的PCR产物去除不干净导致。
大多数所谓的PCR“纯化试剂盒”实际上只是回收产物而不能起到纯化的作用。
对于非特异扩增产物产物肯定是无法去除,而且通常它们不能够完全去除所有的PCR引物,这会造成残留的引物在测序反应过程中参与反应而导致乱峰。
Q-7.如何进行PCR产物纯化?返回顶端A-7.PCR产物首先必须用Agarose胶电泳,将目的条带切割下,然后纯化。
使用凝胶回收试剂盒回收。
产物用ddH2O溶解。
A-8.对于测序用的质粒DNA的要求有哪些?返回顶端A-8. 对于测序用质粒DNA的一般要求:(1) DNA纯度高,1.6~2.0之间,不能有混合模板,也不能含有RNA,染色体DNA,蛋白质等。
(2) 溶于ddH2O中,溶液不能含杂质,如盐类或EDTA等螯合剂,否则将干扰测序反应的正常进行。
Q-9.如何鉴定质粒DNA浓度和纯度?返回顶端A-9.我们使用水平琼脂糖凝胶电泳,并在胶中加入0.5ug/ml的EB,加入一个已知浓度的标准样品。
电泳结束后在紫外灯下比较亮度,判断浓度和纯度。
此方法可以更直接、准确地判断样品中是否含有染色体DNA、RNA等,也可以鉴别抽提的质粒DNA的不同构型。
质粒DNA的3种构型是指在抽提质粒DNA过程中,由于各种原因的影响,使得超螺旋的共价闭合环状的质粒(SC)的一条链断裂,变成开环状(OC)分子,如果两条链发生断裂,就变成线状(L)。
这3种分子有不同的迁移率,通常,超螺旋(SC)迁移速度最快,其次是线状(L)分子,最慢为开环状(OC)分子。
使用紫外分光光度计检测,或者用EB-标准浓度DNA比较法只能检测抽提到的产物中的浓度,甚至由于抽提的质粒DNA中含有RNA、蛋白质、染色体DNA等因素的干扰,浓度检测的数值也是没有多少意义的。
Q-10.对测序引物的要求有哪些?返回顶端A-10.对测序引物的一般要求:(1) 特异性与测序模板结合,不能有多于4个碱基以上的错配现象(2) 不能含有混合碱基(3) 长度17~25碱基(4) 纯度高,最好PAGE纯化(5) 用ddH2O溶解,不要用TE缓冲液溶解。
Q-11.为什么测序引物必须特异地与DNA模板结合?返回顶端A-11.测序引物与待测样品DNA分子只能有一个结合位点是测序成功的关键。
如果测序引物在DNA模板分子上有不只一个的结合位点,将造成测序反应过程中引物链在几个结合位点处同时扩增,反映在测序峰图上将出现双峰或乱峰,无法读取序列。
Q-12.测序结果有很多套峰(出现很多N),还照常收费,为什么?返回顶端A-12.DNA模板上出现二处以上的引物结合位点,或者DNA模板上有严重的重复序列,以及测序引物不纯时, 测序结果便会出现套峰现象(见图4)。
出现这种现象的原因由DNA模板本身或者引物本身所造成,对这些结果(公司保证进行2次以上的测序工作),公司会根据具体情况进行收费(详细见测序结果说明)。
Q-13.为什么用PCR产物测序时,经常会出现套峰现象?返回顶端A-13.PCR产物测序出现套峰现象,一般有以下几种原因:(1) PCR用模板不纯或PCR用引物特异性不好,扩增出的产物除了目的片段外,还有与目的片段长度相近的片段,即使用凝胶电泳也无法分离开,这样的PCR产物测序结果是套峰。
(2) 结构上的原因,造成了PCR产物测序出现套峰的现象。
PolyA/G/C/T以及原因不明的复杂结构的存在,都会出现测序结果套峰的情况。
Q-14.出现套峰的原因是什么?返回顶端A-14.在测序反应中,模板或引物的原因都可能造成套峰的形成,归结其形成原因有以下几点(1) 测序引物在模板上有两个结合位点形成套峰(2) 模板不纯,如果是质粒或是菌液,原因是非单克隆,如果是PCR,原因为非特异性条带(3) 模板序列的特殊结构,如poly结构、发卡结构等(4) 引物降解,引物不纯,或引物的特异性不好Q-15.测序结果不到800 Bases,还照常收费了,为什么?返回顶端A-15.如在DNA样品中的DNA序列分布匀称,没有复杂结构时,正常的测序反应能保证达到800 Bases以上。
但有一些DNA样品立体结构复杂,造成聚合酶延伸反应终止,测序信号突然减弱或消失,或者测序结果出现套峰现象。
出现这些现象的原因由DNA模板本身所造成(公司保证进行2次以上的测序工作)。
对这些结果,公司会根据具体测序情况,进行收费(详细见测序结果说明)。
出现这些情况的原因分析如下:(1) G/C rich、G/C Cluster:这种情况一般表现为测序信号突然减弱或消失(见图1);(2) A、T的连续结构:这种情况一般表现为A、T连续结构后面的测序结果出现套峰(见图2)。
根据文献记载,原因在于聚合酶进行聚合反应时,由于A或T的连续,聚合酶难以识别完整的每个A或T,在某个A或T的后面便开始进行A或T连续结构以后序列的聚合反应(打滑现象),造成测序结果紊乱,出现套峰。
出现这样的情况,建议反向测序。
一般在多少个A或T的后面能出现这种情况呢? 现在还没有这方面的报道。
根据我们的经验,这一情况的出现和A或T的连续结构后面的序列的排列情况有着直接的关系。
有时10多个A或T的连续结构后面便出现套峰,但有时60~70个A或T的连续结构后面的序列也一样可以完整地读出来。
具体情况还有待考证。
一般来说,PCR片段直接测序时,A或T的连续结构后面的序列测序结果都会出现套峰。
原因在于测序时经历了PCR反应及测序反应(测序反应本身也是PCR 反应)二次聚合酶的打滑现象。
(3) 原因不明的复杂结构,测序结果出现突然信号减弱或消失。
从序列上看,DNA碱基排列并无特别异常。
估计是DNA整体出现复杂结构,从某一位置开始聚合酶的聚合反应便无法进行(见图3)。
查看大图Q-16.为什么在测序报告上找不到引物序列?返回顶端A-16.这里分四种情况:(1) 的确找不到测序使用的引物序列。
目前使用的测序方法是在ddNTP上做荧光标记,测序仪通过检测ddNTP上的荧光来读取序列,因为引物本身是不做荧光标记的,所测序列是从引物3' 末端后第一个碱基开始的,所以在测序结果上找不到测序引物的序列。
如果是PCR产物,要想得到PCR引物的序列,可以将PCR 产物进行双链测通或者将PCR产物克隆到载体上,用载体上的引物(注意此引物也不能离插入片段太近)测序(2) 找不到克隆片段的扩增引物。
原因可能是您在构建质粒时采用的工具酶的酶切位点距离您的测序引物太近,由于荧光染料的干扰在序列开始的部分不会十分准确(3) 还有一种可能是您的插入片段的插入方向是反的,这时您不妨找一下您引物的互补序列(4) 存在单引物扩增,有一条引物的特异性不好,有多个结合位点导致只有一条引物参与扩增Q-17.在测序结果上,找不到测序用引物后面的序列,为什么?返回顶端A-17.由于测序仪自身缺陷,紧接引物之后的测序结果信号较弱,一般在测序用引物后面几个至数十个(严重时40~50个)碱基会读不出来(或者读错),请引起注意。
Q-18.怎样使用金斯瑞提供的测序报告?返回顶端A-18.在我们提供的测序报告中,有一份打印的(并用E-mail提供)DNA碱基排列顺序的测序结果。
这个结果是我们根据测序仪的自动分析结果,并经严格确认后打印(并用E-mail提供)的测序结果。
但测序仪有时会发生误读或漏读现象,而我们的工作人员在检查时也没有发现,这时的打印结果(并用E-mail提供)会出现错误。
只有原始的测序彩色波形图才是最正确的,请客户拿到结果后,进行详细确认。
如有不明白的地方,请立即和我们联系,我们会及时确认并进行解决。
Q-19.PCR片段直接测序和PCR片段经克隆后测序的结果有何区别?返回顶端A-19.众所周知,PCR扩增过程中会出现很多错配现象,但不可能所有的错配都发生在同一位置。