测序结果分析教学文案
转录组测序结果解读

转录组测序结果解读
转录组测序是一种重要的基因组学技术,可以帮助科学家了解细胞内基因的表达模式和功能。
在转录组测序实验中,我们可以获得大量的RNA序列数据,通过对这些数据的解读,可以揭示基因在特定生理或病理条件下的表达水平和差异,从而进一步研究基因的调控机制和功能。
首先,转录组测序的结果可以帮助我们了解基因的表达水平。
通过对RNA序列数据进行比对和定量分析,可以计算出每个基因在样本中的表达水平。
这有助于我们确定哪些基因在特定条件下得到了上调或下调,并从中找出具有重要生物学意义的基因。
这些不同表达的基因可能与特定的生理过程、疾病发生和治疗效果等密切相关。
其次,转录组测序还可以帮助我们发现新的基因和转录本。
通过对RNA序列数据进行拼接和注释分析,我们可以发现一些未知的基因和转录本。
这些新的基因和转录本的发现可能会给我们带来对生物学过程的新的认识和思考。
此外,转录组测序结果的解读还包括富集分析和功能注释。
富集分析可以将不同表达的基因进行功能分类,进一步研究基因在特定途径和生物功能中的作用。
功能注释则可以帮助我们预测基因编码的蛋白质的功能和相互作用网络。
综上所述,转录组测序结果的解读是一个复杂但非常重要的工作。
通过对转录组测序数据的分析和解读,我们可以更好地理解基因的表达规律和功能,为生物学研究和医学应用提供有力的支持。
测序结果分析及序列拼接33页文档

பைடு நூலகம்
测序结果分析及序列拼接
46、法律有权打破平静。——马·格林 47、在一千磅法律里,没有一盎司仁 爱。— —英国
48、法律一多,公正就少。——托·富 勒 49、犯罪总是以惩罚相补偿;只有处 罚才能 使犯罪 得到偿 还。— —达雷 尔
50、弱者比强者更能得到法律的保护 。—— 威·厄尔
31、只有永远躺在泥坑里的人,才不会再掉进坑里。——黑格尔 32、希望的灯一旦熄灭,生活刹那间变成了一片黑暗。——普列姆昌德 33、希望是人生的乳母。——科策布 34、形成天才的决定因素应该是勤奋。——郭沫若 35、学到很多东西的诀窍,就是一下子不要学很多。——洛克
植物基因组测序完成结果初步分析报告

植物基因组测序完成结果初步分析报告简介:本报告基于对植物基因组测序完成结果的初步分析,旨在提供对测序数据的解读和分析,以及相关发现和未来研究的建议。
背景:随着高通量测序技术的迅速发展,植物基因组测序成为现代生物学的重要研究领域之一。
植物基因组测序的完成为我们理解植物基因组的结构、功能和进化提供了重要的工具和资源。
本次测序旨在获得某植物的完整基因组序列,为进一步研究该植物的功能基因提供参考。
结果分析:1. 基因组大小估计:通过对测序数据的初步分析,我们得出了该植物的基因组大小估计。
基因组大小是指一个生物体所有基因组成的总长,是评估基因组复杂性和特征的重要指标。
根据我们的分析,该植物预计的基因组大小为XX Mb。
2. 基因注释:我们利用已知的植物基因组数据库和基因预测软件对测序数据进行了基因注释。
通过比对已有的基因序列与我们测序结果的相似性,我们成功注释了一部分的基因,包括编码蛋白质的基因和非编码RNA基因。
同时,我们还发现了一些新的基因,这些新基因可能与该植物在特定环境中的适应性具有重要的联系。
3. 基因家族和表达谱研究:我们进一步对注释的基因进行了家族分析,发现了一些具有重要功能和进化意义的基因家族。
家族分析的结果有助于我们深入理解该植物基因组的起源和进化。
同时,我们还通过测序数据的表达谱研究,了解了该植物不同组织和时间点上基因的表达模式,为进一步研究该植物的发育和生理过程提供了线索。
4. 功能注释和通路分析:我们还对测序结果的基因进行了功能注释和通路分析。
通过比对已知的功能数据库,我们成功注释了一部分基因的功能。
进一步地,通过通路分析,我们发现了一些显著富集的通路以及基因在这些通路中的参与度,有助于我们深入了解该植物的生理和代谢过程。
未来研究建议:1. 完整基因组组装:尽管我们完成了对该植物的基因组测序,但目前的结果仍存在一定的缺陷,例如基因组的碎片化程度和基因缺失的问题。
因此,今后的研究可以通过进一步优化测序方法和使用高级的组装算法来实现完整基因组的测序和组装。
DNA测序结果分析
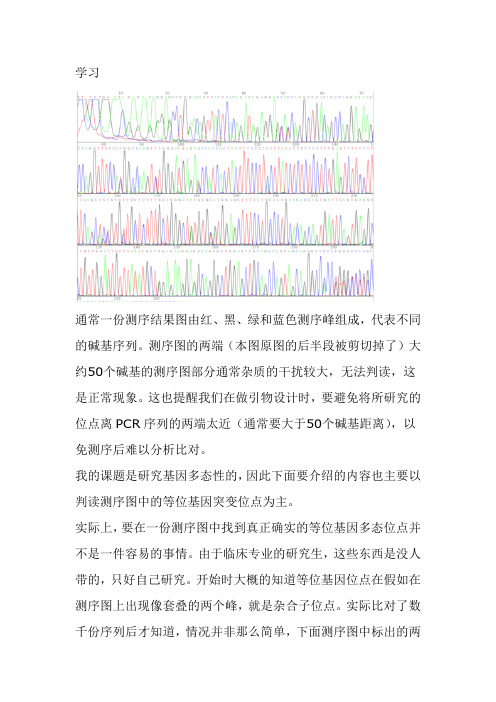
学习通常一份测序结果图由红、黑、绿和蓝色测序峰组成,代表不同的碱基序列。
测序图的两端(本图原图的后半段被剪切掉了)大约50个碱基的测序图部分通常杂质的干扰较大,无法判读,这是正常现象。
这也提醒我们在做引物设计时,要避免将所研究的位点离PCR序列的两端太近(通常要大于50个碱基距离),以免测序后难以分析比对。
我的课题是研究基因多态性的,因此下面要介绍的内容也主要以判读测序图中的等位基因突变位点为主。
实际上,要在一份测序图中找到真正确实的等位基因多态位点并不是一件容易的事情。
由于临床专业的研究生,这些东西是没人带的,只好自己研究。
开始时大概的知道等位基因位点在假如在测序图上出现像套叠的两个峰,就是杂合子位点。
实际比对了数千份序列后才知道,情况并非那么简单,下面测序图中标出的两个套峰均不是杂合子位点,如图并说明如下:说明:第一组套峰,两峰的轴线并不在同一位置,左侧的T峰是干扰峰;第二组套峰,虽两峰轴线位置相同,但两峰的位置太靠近了,不是杂合子峰,蓝色的C峰是干扰峰通常的杂合子峰由一高一略低的两个轴线相同的峰组成,此处的序列被机器误判为“C”,实际的序列应为“A”,通常一个高大碱基峰的前面1~2个位点很容易产生一个相同碱基的干扰峰,峰的高度大约是高大碱基峰的1/2,离得越近受干扰越大。
一个摸索出来的规律是:主峰通常在干扰峰的右侧,干扰峰并不一定比主峰低。
最关键的一点是一定要拿疑似为杂合子峰的测序图位点与测序结果的文本序列和基因库中的比对结果相比较;一个位点的多个样本相比较;你得出的该位点的突变率与权威文献或数据库中的突变率相比较。
通常,对于一个疑似突变位点来说,即使是国际上权威组织大样本的测序结果中都没有报道的话,那么单纯通过测序结果就判定它是突变点,是并不严谨的,因一份PCR产物中各个碱基的实际含量并不相同,很难避免不产生误差的。
对于一个未知突变位点的发现,通常还需要用到更精确的酶切技术。
基因测序报告解读

基因测序报告解读介绍基因测序是一种分析个体基因组的方法,通过对DNA序列进行测定和分析,可以揭示人类基因的组成和变异情况。
基因测序报告是基于个体基因组测序结果的解读分析,能够提供有关个体基因组的丰富信息。
本文将通过一步步的思考,解读基因测序报告。
第一步:基因组信息首先,我们需要了解基因测序报告中提供的基因组信息。
基因组信息通常包括个体的基因型、等位基因频率、基因的功能以及可能的疾病风险等。
通过查阅报告中的基因型信息,我们可以了解个体所携带的基因变异情况。
第二步:常见基因变异接下来,我们需要关注报告中提及的常见基因变异。
基因变异是指个体基因组中与常见基因型不同的碱基序列。
这些变异可能与个体的特定特征或疾病风险相关。
在报告中,常见的基因变异通常会被注明其对应的功能或与之相关的疾病。
第三步:疾病风险评估基于基因测序报告中的基因变异信息,我们可以进一步评估个体的疾病风险。
报告中可能提及一些与基因变异相关的疾病,这些疾病可能包括遗传性疾病、药物反应性等。
通过了解这些信息,我们可以更好地了解个体可能面临的健康风险,并采取相应的预防措施。
第四步:遗传性疾病在报告中,我们也可以找到与遗传性疾病相关的信息。
遗传性疾病是由基因突变引起的疾病,个体携带特定的基因变异可能具有遗传性疾病的风险。
通过了解基因测序报告中与遗传性疾病相关的信息,我们可以提前采取措施,例如进行定期检查、避免特定环境或生活方式等。
第五步:个性化健康管理基于基因测序报告,个体可以进行个性化的健康管理。
通过了解个体基因组中的变异情况,我们可以为个体提供针对其特定基因型的健康建议。
这些建议可能包括特定的饮食和运动指导,以及药物选择和剂量调整等。
结论基因测序报告的解读是了解个体基因组信息的关键步骤。
通过仔细阅读和分析报告中的基因型、基因变异、疾病风险等信息,我们可以更好地了解个体的基因组特征,并为其提供个性化的健康管理建议。
这有助于个体更好地了解自身的遗传特征,预防潜在的疾病,并改善生活质量。
测序结果分析

测序结果分析DNA测序技术已经成为了生物学研究的一项非常重要的工具,能够为我们提供大量的基因信息。
但是,得到大量的基因信息并不意味着我们就可以直接进行基因研究,我们还需要对测序结果进行深入的分析才能够更加准确地了解这些基因的特点和功能。
本文将介绍测序结果分析的基本流程和一些常见的方法。
测序结果分析的基本流程测序结果分析的基本流程可以分为以下几步:1.原始测序数据处理:对测序测序的原始数据进行过滤、去重、修剪等操作,得到高质量的序列数据。
2.序列比对:将样本读取序列与参考序列比对,确定SNP、INDEL、等变异信息。
3.基因注释:对比对结果进行注释,在数据库中查找相关基因的信息,如基因的长度、功能、结构、等等。
4.功能富集分析:对匹配到的基因进行功能富集分析,可以了解到哪些基因和功能在样本间被不同地表达。
常见的测序结果分析方法1. 原始数据处理原始数据处理主要包括数据过滤、去除低质量序列、去除接头等步骤。
之后,我们可以得到高质量的序列数据,用于后续的分析。
2. 序列比对序列比对是将样本测序序列与参考序列比对,比对结果用于寻找SNP、INDEL 等变异。
比对的方法包括全局比对和局部比对,全局比对速度慢但结果较准确,局部比对速度快但可能会产生错误结果。
一般常用的软件有Bowtie、BWA、STAR 等。
3. 基因注释基因注释是对比对结果定位到相应的基因序列上,然后通过与数据库进行比对进行注释。
常用的数据库包括NCBI GenBank、KEGG、UniProt等。
从中可以获得各种相关信息,例如功能、结构、长度等。
4. 功能富集分析功能富集分析是对匹配到的基因进行功能分析,可以了解哪些基因和功能在样本间被不同地表达。
常用的软件包括DAVID(Database for Annotation, Visualization and Integrated Discovery)、GO(Gene Ontology)等,这些数据库可以将富集到的功能直接表示为直线图或散点图等方式,分析结果比较清晰。
PCR引物设计及测序结果分析

PCR技术原理
以拟扩增的DNA分子为模板,以一对分别与 模板5′末端和3′末端互补的寡核苷酸片段为引 物,在DNA聚合酶的作用下,按照半保留复制的机 制沿着模板链延伸直至完成新的DNA合成,重复这 一过程,即可使目的DNA片段得到扩增。
基本原则:
引物与模板的序列要紧密互补 引物与引物之间避免形成稳定的二聚体
或发夹结构 引物不能在模板的非目的位点引发DNA
聚合反应(即错配)。
一般原则:
1. 引物的长度:配对引物的长度一般在1530bp之间比较合适。
尽可能使用两条引物的Tm值相同(最好相差不要超过 5℃)
Tm值的计算:一般的公式:Tm = 4 (G+C) + 2(A+T)
正义链
反义链
基本分子生物学研究手段
PCR PCR反应液体的成分:
PCR循环的程序: PCR原理:
基本分子生物学研究手段
RT-PCR
反转录PCR mRNA---cDNA-----PCR
基本分子生物学研究手段
Cutting by enzymes
基本分子生物学研究手段
Southern Blot
基本分子生物学研究手段
生物软件网下载 安装后,用文本编辑器打开WIN.INI,将vspace=DU
改为vspace=PU便可以使用全部功能。
Oligo primer 3 The Primer Generator NetPrimer
如何使用Primer Premier 5.0
测序结果分析

测序结果的判读测序结果为.abi格式,可用软件chrosmas打开,一种颜色的峰代表一个碱基,峰的高低表信号的强弱。
一个正常的N表示机器没法判读是哪种碱基,原因是:杂峰的信号高于机器默认的值,机器会认为该处有两个峰,因此不能判断确定是哪个峰,需要人工判读。
以下三种情况会出现N:有杂合子,有杂峰,反应已结束。
原因:测序产物纯化不够注意:染料峰位于序列的前100 碱基以内;酒精峰位于序列的220 ~ 320 碱基之间产生的原因是样品或毛细管内有灰尘等固体小颗粒原因:测序反应失败。
解决办法:改进条件,重做反应。
注意两个关键因素:引物与模板之间的比例:3.2 pmol: 200 ng。
模板DNA 的纯度和用量:1.6 ~ 2.0原因:残余的Dye 太多,纯化不够。
有测序反应,但效率低下信号太弱解决办法:纯化充分。
避开引物峰,确定新的分析起点1、PCR产物测序时出现重叠峰问题图1(模板中有碱基缺失,往往是单一位点(1-1)或两个位点(1-2)碱基缺失导致测序结果移码)解决方法:将PCR产物克隆到质粒(如T载体)中挑单克隆测序,或将PCR产物进行PAGE 纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序。
问题图2(PCR产物不纯,含部分序列一致的两种以上的片段,长度不一)解决方法:主要原因是PCR产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE纯化2、克隆测序时出现峰形重叠原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液再次测序。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
测序结果的判读
测序结果为.abi格式,可用软件chrosmas打开,一种颜色的峰代表一个碱基,峰的高低表信号的强弱。
一个正常的N表示机器没法判读是哪种碱基,原因是:杂峰的信号高于机器默认的值,机器会认为该处有两个峰,因此不能判断确定是哪个峰,需要人工判读。
以下三种情况会出现N:有杂合子,有杂峰,反应已结束。
原因:测序产物纯化不够
注意:染料峰位于序列的前100 碱基以内;酒精峰位于序列的220 ~ 320 碱基之间
产生的原因是样品或毛细管内有灰尘等固体小颗粒
原因:测序反应失败。
解决办法:改进条件,重做反应。
注意两个关键因素:引物与模板之间的比例:3.2 pmol: 200 ng。
模板DNA 的纯度和用量:1.6 ~ 2.0
原因:残余的Dye 太多,纯化不够。
有测序反应,但效率低下信号太弱
解决办法:纯化充分。
避开引物峰,确定新的分析起点
1、PCR产物测序时出现重叠峰
问题图1(模板中有碱基缺失,往往是单一位点(1-1)或两个位点(1-2)碱基缺失导致测序结果移码)
解决方法:将PCR产物克隆到质粒(如T载体)中挑单克隆测序,或将PCR产物进行PAGE 纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序。
问题图2(PCR产物不纯,含部分序列一致的两种以上的片段,长度不一)
解决方法:主要原因是PCR产物没有纯化,含有部分序列一致的两种以上长度不一的片段,将PCR产物进行PAGE纯化(至少琼脂糖充分电泳后切胶纯化)后再进行测序,便可解决。
问题图3(测序引物有碱基缺失)
测序引物有碱基缺失(一般是引物的5'端缺失),和模板的碱基缺失即图1有些类似,所不同的是模板碱基缺失一般是在一段正常测序序列后才出现移码,而引物碱基缺失的话,则从测序一开始就出现移码,表面在图形上便是一开始就是严重的峰形重叠。
解决方法:重新合成引物,或将引物进行PAGE纯化
2、克隆测序时出现峰形重叠
原因:所挑选的重组子不是单克隆,所提供的测序用质粒中含有两种以上插入片段不同的质粒;或是是送测序的菌液污染
解决方法:重新挑单克隆的菌落(划线分离单菌落),提质粒或送菌液再次测序。
3、样品有杂合/突变位点
模板中有杂合型突变,也就说模板本身在这个位点出现突变;或者是从基因组中扩增出来的杂合位点。
如果模板有杂合(突变或缺失),那么测序图形中其他的位點一般都是單一的峰形,然后突然在某一個位點出現重叠峰(如图中箭頭所示)。
解决方法:建议将DNA片段克隆到载体再测序。
4、polyA/T和C/Gcluster导致的套峰和测序信号衰减
RACE测序时经常遇到图4-1和图4-4的情形,解决方法:从另一端测序;或者构建载体进行测序。
5、基因中含有重复序列
可能的原因:样品中含有重復序列导致的测序结果和PolyA/T的结果一样,会导致Frame 滑动,较短的重復序列会导致测序结果出现移码;而较长的重復序列会使定序信号衰减。
解决办法:反向测序有时能够顺利的通过重復序列区域(但不是一定都能够),通过多次的测序结果比对,拼接可以得到全序列结果。