– Analysis of Microarray images
meta-analysis-元分析

7
Rauch A, Wiklund J, Lumpkin G T, et al. Entrepreneurial Orientation and Business Performance: An Assessment of Past Research and Suggestions for the Future[J]. Entrepreneurship Theory & Practice, 2009, 33(3):761–787.
15
Jain R K. Entrepreneurial Competencies A Meta-analysis and Comprehensive Conceptualization for Future Research[J]. Vision the Journal of Business Perspective, 2011, 15(2):127-152.
3
Dan R D, Daily C M, Certo S T, et al. Meta-Analyses of Financial Performance and Equity: Fusion or Confusion?[J]. Academy of Management Journal, 2003, 46(1):13-26.
16
Samaraweera M, Gelb B D. Formal salesforce controls and revenue production: a meta-analysis[J]. Journal of Personal Selling & Sales Management, 2015, 35(1):23-32.
anderson-基因芯片课程-ma00b

INTRODUCTION TO MICROARRAYS
1
Lecture 0: Outline and Details
Welcome!
• Where are we? • Why are we here? • Who are we? • What are we going to tell you about, and when? • What do we assume you’re familiar with? • Where and when can you find ??? • What are we going to ask of you?
c Copyright 2009 Keith A. Baggerly and Brad M. Broom
GS01 0163: ANALYSIS OF MICROARRAY DATA
INTRODUCTION TO MICROARRAYS
3
So, why are we here?
We want to learn about microarrays.
Optional: Simon RM, et al. Design and Analysis of DNA Microarray Investigations. Springer-Verlag, New York, 2003.
Optional: Dalgaard P. Introductory Statistics with R. Springer-Verlag, New York, 2002.
c Copyright 2009 Keith A. Baggerly and Brad M. Broom
GS01 0163: ANALYSIS OF MICROARRAY DATA
MDH1介导苹果酸-天冬氨酸NADH穿梭维持胎儿肝造血干细胞的活性水平。(Sonarsignal)
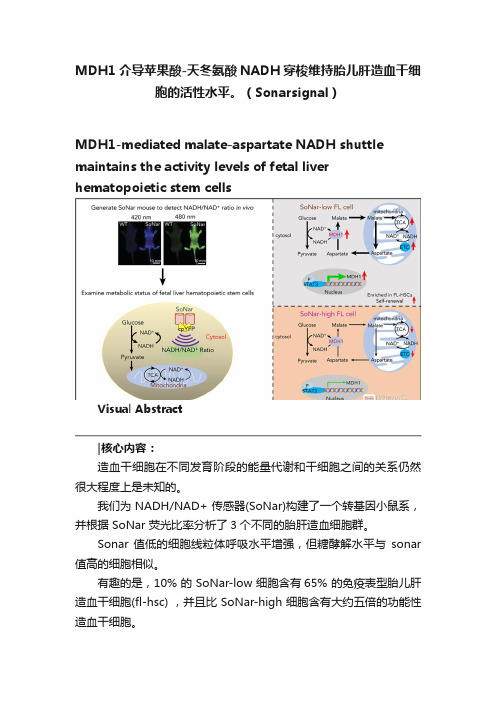
MDH1介导苹果酸-天冬氨酸NADH穿梭维持胎儿肝造血干细胞的活性水平。
(Sonarsignal)MDH1-mediated malate-aspartate NADH shuttle maintains the activity levels of fetal liver hematopoietic stem cellsVisual Abstract造血干细胞在不同发育阶段的能量代谢和干细胞之间的关系仍然很大程度上是未知的。
我们为 NADH/NAD+ 传感器(SoNar)构建了一个转基因小鼠系,并根据 SoNar 荧光比率分析了3个不同的胎肝造血细胞群。
Sonar 值低的细胞线粒体呼吸水平增强,但糖酵解水平与sonar 值高的细胞相似。
有趣的是,10% 的 SoNar-low 细胞含有65% 的免疫表型胎儿肝造血干细胞(fl-hsc) ,并且比 SoNar-high 细胞含有大约五倍的功能性造血干细胞。
Sonar 能够敏感地监测体内外造血干细胞能量代谢的动态变化。
STAT3通过反式激活 MDH1维持苹果酸-天冬氨酸 NADH 的穿梭活性和 HSC 的自我更新和分化。
我们揭示了一个意想不到的fl-hsc 代谢程序,为造血干细胞或其他类型干细胞的代谢研究提供了一个强有力的遗传工具。
原文摘要:The connections between energy metabolism and stemness of hematopoietic stem cells (HSCs) at different developmental stages remain largely unknown.We generated a transgenic mouse line for the genetically encoded NADH/NAD+ sensor (SoNar) and demonstrate that there are 3 distinct fetal liver hematopoietic cell populations according to the ratios of SoNar fluorescence.SoNar-low cells had an enhanced level of mitochondrial respiration but a glycolytic level similar to that of SoNar-high cells. Interestingly, 10% of SoNar-low cells were enriched for 65% of total immunophenotypic fetal liver HSCs (FL-HSCs) and contained approximately fivefold more functional HSCs than their SoNar-high counterparts.SoNar was able to monitor sensitively the dynamic changes of energy metabolism in HSCs both in vitro and in vivo.Mechanistically, STAT3 transactivated MDH1 to sustain the malate-aspartate NADH shuttle activity and HSC self-renewal and differentiation.We reveal an unexpected metabolic program of FL-HSCs and provide a powerful genetic tool for metabolic studies of HSCs or other types of stem cells.Key Points•FL-HSCs mainly use oxidative phosphorylation but with normal glycolysis, as indicated by a highly responsive NADH/NAD+ sensor.•FL-HSC activities are tightly regulated by the STAT3/MDH1-mediated malate-aspartate NADH shuttle.Subjects:Hematopoiesis and Stem CellsTopics:aspartate, fetus, fluorescence, liver, malates, mice, transgenic, mitochondria, nicotinamide adenine dinucleotide (nad), stat3 protein, hematopoietic stem cellsIntroductionHematopoietic stem cells (HSCs) originate from the aorta-gonad-mesonephros region1 and migrate into the fetal liver (FL) and undergo dramatic expansion,2,3 gradually localizing to and residing in the bone marrow niche after birth.4HSCs can self-renew to maintain the stem cell pool and generate all downstream progenitors and terminally differentiate into multiple lineages.5,6Increasing evidence indicates that the metabolic state is tightly connected to HSC activity.7-9Adult HSCs preferentially undergo glycolysis, rather than oxidative phosphorylation, in the hypoxic niche,7,10,11 which is extensively regulated by several signaling pathways, including HIF1A,12 MYC,13 PDK,14 DLK-GTL2,15 and vitamin A–retinoic acid signaling.16We have also shown that both murine and human HSCs adopt a glycolytic metabolic profile under certain conditions andthat this profile is fine-tuned by MEIS1/PBX1/HOXA9/HIF1A signaling pathways.17-19Interestingly, recent studies have suggested that adult HSCs also have high mitochondrial mass and enhanced dye efflux but possess limited respiratory and turnover capacity,20 which indicates that mitochondria are likely required for the function of adult HSCs, as evidenced by the fact that FOXO3 serves as a regulator to couple mitochondrial metabolism with HSC homeostasis.21The metabolic profiles of FL-HSCs and the effects of metabolism on HSC function, however, remain largely unknown.FL-HSCs undergo rapid division/expansion, conceivably through an increased demand on energy sources compared with that needed by adult HSCs, which are usually maintained in a relatively quiescent state.It is also possible that distinct microenvironments in different hematopoietic organs may affect the metabolism of HSCs.Interestingly, a recent report showed that loss of Rieske iron-sulfur protein, a mitochondrial complex III subunit, impairs the quiescent status of adult HSCs and the differentiation capacity of FL-HSCs.22FL-HSCs seem to have increased expression levels of many mitochondrial respiration–related genes, although whether metabolic status determines the cell fate of FL-HSCs remains unknown.23Results from previous studies indicate that mitochondrial activity may play a role in HSCs in the FL stage, although the detailed metabolic profiles and their underlying mechanisms await further investigation.Because of limitations in the availability of HSCs, moststudies related to the nutrient metabolism of HSCs have depended heavily on flow cytometric analysis with MitoTracker dyes, TMRE, and DCFDA to determine mitochondrial mass, membrane potential, and ROS level, respectively.Improved techniques have been used to measure several metabolic features of HSCs, such as oxygen consumption and lactate generation9,24 ; however, these studies may not directly reflect the true extent of glycolysis, oxidative phosphorylation, or other metabolic processes in HSCs.Recent studies have provided interesting evidence showing that it is feasible to perform a metabolomic analysis with fewer than 104 HSCs to explore the metabolic networks of different types of nutrients.25Nevertheless, it remains difficult to detect all of the metabolites sensitively with a limited number of HSCs using conventional metabolomic analysis.Few tools are available for real-time imaging of metabolic states in live HSCs, either in vitro or in vivo. Therefore, alternative approaches, such as metabolite biosensors, are required for the direct, precise, and real-time detection of subtle changes in nutrient metabolism in HSCs.Recently, we developed a highly responsive NADH/NAD+ sensor, called SoNar,26 which was designed by inserting cpYFP into the NAD(H)-binding domain of T-Rex.SoNar shows distinct fluorescence responses to NADH and NAD+.Inside the cell under physiological conditions, the total intracellular pool of NAD+ and NADH in the range of hundreds of micromolars27-30 far exceeds the dissociation constants of SoNar for NAD+ (K d, 5.0 μM) and NADH (K d, 0.2 μM);thus, the sensor would be occupied by either NAD+ or NADH molecules, and its steady-state fluorescence would report the NAD+/NADH ratio rather than the absolute concentrations of either of the 2 nucleotides according to equilibrium thermodynamics.26In addition, SoNar fluorescence is intrinsically ratiometric (比率计), with 2 excitation wavelengths, and its fluorescence excited at 420 (or 405) and 485 nm shows opposing responses to ligand binding.26,31This ratiometric property of a sensor is highly desired for quantitative imaging in live cells and in vivo,32,33 because it eliminates the differences in instrumental efficiency, environmental effects, and probe concentration, enabling it to be widely used in different biological samples.The SoNar sensor has a 15-fold (or 1500%) dynamic range, enabling us to measure the cytosolic NAD+/NADH ratio from 0.8 to 2000.26,31,34Interestingly, SoNar has many desirable properties that make it an ideal sensor; it has a rapid response, high sensitivity, intense fluorescence, and large dynamic range, and it is capable of reporting subtle perturbations in many pathways affecting energy metabolism, including glycolysis and mitochondrial respiration.We generated SoNar transgenic mice and examined the metabolic profiles of FL-HSCs and their contributions/connections to cell fate determinations, as well as the underlying mechanisms governing FL-HSC function.参考文献:/10.1182/blood.2019003940HEMATOPOIESIS AND STEM CELLS| JULY 30, 2020----------------------------------------------------------------------------------------------------------------------------MethodsMiceSoNar DNA consists of the sequence of cpYFP, truncated T-Rex (78-211), and the linkers between them.26 Its coding gene (1.2 kbp) is much smaller than those of the first-generation NADH sensors Peredox (2.8 kbp) and Frex35 (1.8 kbp). For transgenic studies, smaller is better for the expression of the sensor in cells and in vivo.31 To generate SoNar transgenic mice, SoNar DNA was cloned into the pCAG vector with chicken β-actinpromoter. The targeting construct was linearized, purified, and microinjected into FVB blastocysts. SoNar DNA was randomly incorporated into the genome and determined by polymerase chain reaction (PCR) assay. Messenger RNA (mRNA) and protein expressions of SoNar in different tissues of SoNar mice were further evaluated by both reverse transcription PCR (RT-PCR) and fluorescence microscopy. The resulting chimeric mice were bred with FVB mice to obtain germ line transmission. These mice were next backcrossed with a C57BL/6 CD45.2 background, and germ line transmission was checked by PCR and flow cytometry. Heterozygote transgenic SoNar mice were used for most of the experiments in the current study. C57BL/6 CD45.2 mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. CD45.1 mice were provided by Dr Jiang Zhu at Shanghai Jiao Tong University School of Medicine. All animal experiments were conducted according to the Guidelines for Animal Care at Shanghai Jiao Tong University School of Medicine. All these materials, including SoNar sensor, are available upon request.In vivo imaging of SoNar transgenic miceGenotyping, mRNA expression, and histology of SoNar transgenic miceCompetitive reconstitution assayMetabolic imaging and quantification of cytosolicNAD+/NADH ratio in living cellsReal-time metabolic imaging in the BM nicheFlow cytometryUltrahigh-performance LC–qTOF–MS analysisMicroarray and quantitative RT-PCRMetabolic analysisOxygen consumption rate (OCR) and extracellularacidification rate were determined in CD45.2+ SoNar-high and -low FL hematopoietic cells with the XF Cell Mito Stress Test Kit (Seahorse #103015-100) and XF Glycolysis Stress Test Kit (Seahorse #103020-100) according to the manufacturer’s instructions using a Seahorse XF96 analyzer. In brief, for the OCR analysis, 3 × 105 SoNar-high and -low FL hematopoietic cells were incubated in the 37°C carbon dioxide–free incubator in 175 μL of assay medium (XF Base Medium with 2 mM of glutamine, 1 mM of pyruvate, and 10 mM of glucose [pH, 7.4]; 37°C); 1.5 μM of oligomycin, 2 μM of FCCP, and 0.5 μM of rotenone/antimycin A were loaded in injection ports A, B, and C, respectively. For the detection of extracellular acidification rate, 3 × 105 CD45.2+ SoNar-high and -low FL hematopoietic cells were incubated in the 37°C carbon dioxide–free incuba tor in 175 μL of assay medium (XF Base Medium with 1 mM of glutamine [pH, 7.4]; 37°C); 10 mM of glucose, 1.5 μM of oligomycin, and 100 mM of 2-DG were loaded into injection ports A, B, and C, respectively. ATP level was analyzed using the ATP Bioluminescence Assay Kit HS II (Roche) according to the manufacturer’s protocol, and data were normalized to cell count. T o analyze the mitochondrial DNA (mtDNA) copy numbers, total genomic DNA was extracted from the indicated cells for comparing the copies of the mitochondrial-specific mt-ND4 gene with those of the nuclear B2m gene. The primer sequences used are shown in supplemental Table 1.For intracellular and extracellular pyruvate and lactate assays, the extracts were prepared from 5 × 106 CD45.2+ FL hematopoiet ic cells using 300 μL of ice-cold 0.5-M perchloric acid for each sample. Extracts were centrifuged at 10 000 g for 5 minutes at 4°C, and the supernatant was neutralized with 5 M ofKOH and centrifuged at 10 000 g for 5 minutes at 4°C. The supernatant was removed for assay. For the pyruvate assay, 180 μL of assay buffer (100 mM of potassium chloride [pH, 6.7], 1 mM of EDTA, 0.1% bovine serum albumin, 10 μM of flavin adenine dinucleotide, 0.2 mM of thiamine pyrophosphate, 0.5 U of pyruvate oxidase, 0.2 U of h orseradish peroxidase, and 50 μM of AmplexRed) was added to a 96-well plate containing 20 μL of the cell extract or medium containing extracellular pyruvate. Changes in fluorescence were measured every 30 seconds for 15 minutes at 37°C by a Synergy 2 Multi-Mode Microplate Reader with an excitation filter of 530 BP 40 nm and emission filter of 590 BP 35 nm at 37°C. Calibration experiments were performed with 20 μL of pyruvate standards (0, 10, 20, 40, 60, 100, and 200 μM per well). For the lactate assay, 180μL of assay buffer (PBS [pH, 7.4], 0.1% bovine serum albumin, 500 μM of NAD+, 0.5 U of lactate dehydrogenase (LDH), 0.2 U of diaphorase, and 10 μM of resazurin) was added to a 96-well plate containing 20 μL of the cell extract or medium containing extracellular lactate. Changes in fluorescence were measured every 30 seconds for 15 minutes at 37°C by a Synergy 2 Multi-Mode Microplate Reader with an excitation filter of 540 BP 25 nm and emission filter of 590 BP 35 nm at 37°C. Calibration experiments were performed with 20 μL of lactate standards (0, 10, 20, 40, 60, 100, and 200 μM per well). All samples were diluted to fit within the range of the standard curve and run in triplicate. For the evaluation of NADH/NAD+ ratios by a biochemical assay, 1 million SoNar-high and -low FL hematopoietic cells were sorted from E14.5 FLs and subjected to measurement of NADH/NAD+ level using a commercially available NADH/NAD+ assay kit (Sigma #MAK037) according to the manufacturer’s instructions.Immunoblot analysisSingle cell colony formingThe single CD45.2+ SoNar-high or -low FL hematopoietic cell was freshly isolated and plated onto a 35-mm poly-D-lysine hydrobromide-coated glass-bottom dish (Cellvis) and cultured in Stemspan serum-free medium (Stemcell Technologies) containing 10 ng/mL of murine SCF and 10 ng/mL of murine TPO (Peprotech) for 96 hours. The ratios of daughter cells derived from a single parent cell were recorded with excitation at 405 and 488 nm using Nikon A1 confocal microscopy and analyzed with Image J software.Luciferase reporter assaysThe luciferase reporter vector pGL4.27 containing the Mdh1 promoter was constructed to identify transcriptional activation of Mdh1 by STAT3. Indicated doses of pLVX-Stat3 (or negative control vector) plasmid along with pGL4.27-mdh1 promoter vector were cotransfected into 293T cells. Luciferase activities were measured according to the manufacturer’s instructions (Promega #E1910) by using a luciferase reporter system (GloMax Multi Instrument) 24 hours after transfection. ChIP assaysChromatin immunoprecipitation (ChIP) assays were performed using the ChIP Assay Kit (Beyotime #P2078). Briefly, 293T cells were overexpressed with pGL4.27-mdh1 promoter vector and STAT3 (with Strep II tag) crosslinked with 1% formaldehyde (S igma) at 37°C for 10 minutes, and precleared DNA was then used for immunoprecipitation with 4 mL of anti–Strep II antibody (Genescript) or rabbit control immunoglobulin G (CST) at 4°C overnight. For the sample input, 1% of the sonicated pre-cleared DNA was purified at the same time withthe precipitated immune complex. The ChIP samples were purified by the Gel and PCR Clean Up Kit (Necleospin). The STAT3-binding sequence was amplified by semiquantitative PCR using primers specific for the Mdh1 promoter region as listed in supplemental Table 1.Methylation-specific PCR assayGenomic DNA was extracted from 500 000 CD45.2+ SoNar-high and -low FL hematopoietic cells using a DNA extraction kit (Generay Biotech #GK0122). The promoter methylation status of MDH1 was determined by sodium bisulfate to convert unmethylated (but not methylated) cytosine to uracil, followed by analysis by methylation-specific PCR to amplify specifically either methylated or unmethylated DNA using the Zymoresearch kit (EZ DNA Methylation-Direct Kit D5020) according to the manufacturer’s instruction. The methylation-specific PCR primers are listed in supplemental Table 1.Statistical analysisStatistical analysis was performed using GraphPad and SPSS software (version 19.0). Data are represe nted as mean ± standard error of the mean. n represents the number of independent experiments or the number of cells or mice per group from independent experiments. All experiments were performed independently 3 to 5 times. Data were analyzed with a Student t test (2 tailed), 1-way analysis of variance with Tukey’s multiple comparison test, or 2-way analysis of variance with Sidak’s multiple comparison test according to the experimental design, and statistical significance was set at P < .05.Establishment of pan-tissue SoNar transgenic mice.Figure 2.SoNar indicates metabolically distinct populations of FL hematopoietic cells.Figure 3.SoNar-low FL hematopoietic cells exhibit similar glycolytic but enhanced mitochondrial activity compared with SoNar-high cells.Figure 4.Functional HSCs are enriched in SoNar-low FLhematopoietic cells.Figure 5.FL-HSCs respond differently to AOA stimulation in the BM niche compared with adult HSCs.Figure 6.MDH1 enhances the malate-aspartate NADH shuttle and decreases NADH/NAD+ level in SoNar-low FL-HSCs.Figure 7.STAT3 transactivates Mdh1 expression to maintain FL-HSC activities.。
STEM分析

ularly clinical studies, the availability of biological material can limit the number of time points collected. Thus, even if the price of microarray experiments were to go down short time series expression experiments would remain prevalent. In this paper we introduce the Short Time-series Expression Miner (STEM), the first software application designed specifically for the analysis of short time series gene expression datasets (3–8 time points). Data from short time series gene expression experiments poses
Page 1 of 11
(page number not for citati间序列指的 是涉及的时 间节点比较 少
Derepression of Neuronal Inhibitor due to miRNA Dysregulation in Schizophrenia-Related Microdeletion

Derepression of a Neuronal Inhibitor due to miRNA Dysregulation in a Schizophrenia-Related MicrodeletionBin Xu,1,3,5Pei-Ken Hsu,3,4,5Kimberly L.Stark,1,3Maria Karayiorgou,1,*and Joseph A.Gogos2,3,*1Department of Psychiatry2Department of NeuroscienceCollege of Physicians and Surgeons,Columbia University,1051Riverside Drive,New York,NY10032,USA3Department of Physiology and Cellular Biophysics,College of Physicians and Surgeons4Integrated Program in Cellular,Molecular,and Biophysical StudiesColumbia University,630West168th Street,New York,NY10032,USA5These authors contributed equally to this work*Correspondence:mk2758@(M.K.),jag90@(J.A.G.)/10.1016/j.cell.2012.11.052SUMMARY22q11.2microdeletions result in specific cognitive deficits and schizophrenia.Analysis of Df(16)A+/Àmice,which model this microdeletion,revealed abnormalities in the formation of neuronal dendrites and spines,as well as altered brain microRNAs. Here,we show a drastic reduction of miR-185,which resides within the22q11.2locus,to levels more than expected by a hemizygous deletion,and we demon-strate that this reduction alters dendritic and spine development.miR-185represses,through an evolu-tionarily conserved target site,a previously unknown inhibitor of these processes that resides in the Golgi apparatus and shows higher prenatal brain expres-sion.Sustained derepression of this inhibitor after birth represents the most robust transcriptional disturbance in the brains of Df(16)A+/Àmice and results in structural alterations in the hippocampus. Reduction of miR-185also has milder age-and region-specific effects on the expression of some Golgi-related genes.Ourfindings illuminate the contribution of microRNAs in psychiatric disorders and cognitive dysfunction.INTRODUCTIONThe identification of a widespread role of chromosomal microde-letions and microduplications(copy number variants or CNVs)in determining susceptibility to psychiatric disorders such as schizophrenia(SCZ),as well as neurodevelopmental disorders such as autism and intellectual disability,represents a shift in our understanding of the genetic architecture of these disorders and highlights the pervasive contribution of rare and highly pene-trant structural mutations(Karayiorgou et al.,1995;Morrow, 2010;Rodriguez-Murillo et al.,2012).Along these lines,a strong link has been established between microdeletions in chromo-some22q11.2,cognitive dysfunction and psychiatric disorders, especially SCZ(Karayiorgou et al.,1995,2010;Xu et al.,2008). Understanding how the genes disrupted by this deletion contribute to the ensuing psychiatric and cognitive phenotypes will provide important mechanistic insights and guide analysis of other pathogenic mutations(Arguello and Gogos,2006, 2010,2012;International Schizophrenia Consortium,2008; Karayiorgou et al.,2010).By using chromosomal engineering,we generated a mouse model carrying a hemizygous1.3Mb chromosomal deficiency on mouse chromosome16(Df(16)A),which is syntenic to the 1.5Mb22q11.2microdeletion(Stark et al.,2008).Analysis of Df(16)A+/Àmice showed abnormalities in dendritic morphogen-esis and formation of dendritic spines of hippocampal pyramidal neurons both in culture and in vivo(Mukai et al.,2008;Stark et al., 2008).Such changes may account,at least in part,for the regional decreases in gray matter volumes observed in some 22q11.2deletion carriers(Bearden et al.,2009;Chow et al., 2002)and may ultimately lead to altered information processing. Analysis of the Df(16)A+/Àstrain also provided compelling evidence that the22q11.2microdeletion results in abnormal pro-cessing of brain microRNAs(miRNAs),a class of small noncod-ing RNAs that regulate the stability and translation of mRNAs (Fineberg et al.,2009;Kosik,2006;Schratt,2009;Xu et al., 2010),implicating miRNA dysregulation in the pathogenesis of psychiatric disorders and cognitive dysfunction.One gene dis-rupted by the22q11.2microdeletion is DGCR8,a component of the‘‘microprocessor’’complex that is essential for miRNA production(Tomari and Zamore,2005).Dgcr8haploinsufficiency results in the downregulation of a specific subset of mature miRNAs and contributes to alterations found in Df(16)A+/Àmice (Fe´nelon et al.,2011;Stark et al.,2008).miRNA dysregulation likely accounts for a fraction of the transcript misexpression in the brains of Df(16)A+/Àmice(Stark et al.,2008),but direct targets have not been reported.Here,we highlight an important component of this dysregulation and identify a previously un-characterized gene with prenatal expression bias as amajor262Cell152,262–275,January17,2013ª2013Elsevier Inc.miRNA target mediating the effects of the 22q11.2microdeletion on neuronal maturation and connectivity.RESULTSA Drastic Reduction of miR-185Levels in Df(16)A +/ÀMiceStudies in the Df(16)A +/Àmouse strain have shown that the 22q11.2microdeletion results in abnormal processing of a specific subset of brain miRNAs due to the removal of one copy of the Dgcr8gene causing a decrease in its expression in the adult brain (Stark et al.,2008)as well as in early development (Figure S1A available online).It is noteworthy that,in addition to Dgcr8,the 22q11.2microdeletion and the equivalent mouse deficiency remove one copy of a miRNA gene (miR-185)located within the minimal 1.5Mb 22q11.2critical region (Figure 1A).In situ hybridization assays indicated that miR-185is expressed in several brain regions such as hippocampus (HPC)and cortex (Figure 1B).qRT-PCR analysis showed that expression of miR-185is dramatically reduced by $70%–80%in both HPC(p <10À6)and prefrontal cortex (PFC;p <10À11)of adult Df(16)A +/Àmice as compared to wild-type (WT)littermates (Figures 1C and 1D).This reduction was also observed at earlier develop-mental stages (embryonic day 17[E17]and postnatal day 6[P6])(Figure S1B).miR-185also showed a more modest decrease in Dgcr8+/Àmice ($20%in HPC;p <0.05;Figure 1E),suggesting that the severe reduction of mature miR-185expression in Df(16)A +/Àmice may be due to a combined effect of hemizygos-ity of the miR-185gene and impaired maturation of the pri-mir-185transcript produced from the remaining copy,due to the reduction in Dgcr8levels.Such a large reduction in expression of a resident gene to levels greater than expected by the 50%decrease in gene dosage is unique among genes affected by the 22q11.2microdeletion.A Primary Transcriptional Consequence of 22q11.2Genomic LossPrevious microarray analysis of adult Df(16)A +/Àmice revealed genome-wide alterations of transcriptional programs in the HPC and PFC (Stark et al.,2008).We extendedexpressionFigure 1.Drastic Reduction of mir-185Expression in Df(16)A +/ÀMice(A)Schematic diagram showing the 1.5Mb 22q11.2critical region and the syntenic mouse locus.The 1.5Mb deletion is mediated by low copy repeat sequences LCR-A and LCR-B (illustrated as black boxes).Dgcr8and miR-185(hosted in the intron of the 22orf25gene in human and the D16H22S680E gene in mouse)are highlighted in red.Chr22,chromosome 22;Chr16,chromosome 16.(B)Expression of miR-185mRNA in HPC and cortex as shown by in situ hybridization in coronal brain sections using an antisense miR-185probe.An antisense U6probe and a scramble probe were used as positive and negative controls,respectively.Images were taken at either 43(left)or 103(right)magnification.(C–E)miR-185expression levels in HPC (C)or PFC (D)of Df(16)A +/À(n =7for mutant,n =9for WT)and in HPC (E)of Dgcr8+/À(n =10for mutant and WT),as assayed by qRT-PCR.Expression levels in mutant mice were normalized to their respective WT littermates.Results are expressed as mean ±SEM.*p <0.05,**p <0.01,and ***p <0.001(Student’s t test).See also Figure S1.Cell 152,262–275,January 17,2013ª2013Elsevier Inc.263Figure2.2310044H10Rik Is Robustly Upregulated in the Brain of Df(16)A+/ÀMice(A)Changes in gene expression in the PFC(upper panel)or HPC(lower panel)of Df(16)A+/Àand WT littermate control mice at E16,P6,and adulthood(n=10each group).Volcano plot of the p values and the corresponding relative expression of each gene are shown.Light blue dots indicate genes within Df(16)A deficiency, light green dots indicate upregulated miRNA-containing transcripts,and red dots indicate probe sets representing Mirta22.(B)Top ten protein-encoding genes that show significant upregulation in the PFC(upper panel)or HPC(lower panel)of Df(16)A+/Àand WT littermate mice at E16, P6,and adulthood.Mirta22is highlighted in red.(legend continued on next page)264Cell152,262–275,January17,2013ª2013Elsevier Inc.profile analysis of these two brain regions to two earlier develop-mental stages,E17and P6.Only one gene,2310044H10Rik,was consistently significantly upregulated(in at least two of the three developmental stages examined and in at least one of the two brain areas tested).Indeed,2310044H10Rik was among the top upregulated genes in both postnatal stages examined (Figures2A and2B).Notably,no significant difference in 2310044H10Rik expression was found in either frontal cortex or HPC at E17(Figures2A and2B).Importantly,there is no known miRNA within or surrounding this genomic locus,sug-gesting that the upregulation is not due to impaired processing of overlapping pri-miRNA transcripts.In independent experiments,we attempted to distinguish primary versus secondary gene targets of the22q11.2microde-letion by looking for genes whose expression changes in oppo-site direction as a result of genomic losses or gains in this locus. Such genes are likely to represent primary targets and direct transcriptional readouts of the underlying copy number imbal-ances(Chahrour et al.,2008).We compared the PFC and HPC gene expression profiles in mice carrying a deletion or duplica-tion at the22q11.2syntenic mouse locus using as reference compound heterozygous mice balanced for copy number (see Extended Experimental Procedures;Figure S2).We identi-fied a number of inversely altered transcripts in either PFC or HPC(p<0.001;Extended Experimental Procedures;Table S1),in addition to the transcripts from the22q11.2region.As expected,the majority of the identified transcripts are pri-miRNA forms.Only12transcripts were significantly misregu-lated in a reciprocal manner in both PFC and HPC(Table S2). Among them,2310044H10Rik is the only gene with protein-coding potential.Taken together,our expression profiling highlighted the misre-gulation of2310044H10Rik as a major consequence of the 22q11.2genomic imbalances at the transcriptome level.We confirmed the pattern of2310044H10Rik upregulation in both PFC and HPC by TaqMan qRT-PCR(PFC:E17,20%,p=0.24; P6,59%,p<0.01;Adult,76%,p<10À6;HPC:E17,20%,p= 0.16;P6,50%,p<0.05;Adult,38%,p<0.05;Figures2C and 2D).This analysis revealed a profile of temporal regulation with prenatal expression bias where levels of2310044H10Rik rapidly decline during thefirst week after birth and remain constantly low thereafter,as well as a corresponding pattern of misregula-tion in Df(16)A+/Àmice where prenatally elevated expression persists throughout postnatal and adult life.Increased brain expression of2310044H10Rik is recapitulated in Df(16)A+/Àprimary neurons(Figure2E).2310044H10Rik Is a Major Downstream Effectorof miRNA DysregulationNotably,2310044H10Rik mRNA levels were also elevated in Dgcr8+/Àmice(HPC:30%,p<0.05;PFC:24%,p<0.05;Fig-ure S3A),suggesting that upregulation may be due to miRNA dys-regulation.Indeed,two miRNA target site prediction programs, TargetScan(Grimson et al.,2007)and mirDB(Wang,2008),report that the30UTR of2310044H10Rik contains binding sites of miRNAs shown to be affected in Df(16)A+/Àmice by microarray profiling(Stark et al.,2008).Specifically,mirDB predictedfive such miRNAs with binding sites in the30UTR of2310044H10Rik including miR-185and miR-485,whereas TargetScan predicted 13miRNA sites,including sites for miR-185,miR-485,miR-491, and miR-224.Notably,both programs predicted sites for miR-185and miR-485(Figure3A,red rectangles).Because increased brain expression of2310044H10Rik is recapitulated in Df(16)A+/Àprimary neurons(Figure2E),wefirst used primary neurons to determine whether endogenous 2310044H10Rik expression is actually under the control of miR-185.To examine the effect of miR-185overexpression on 2310044H10Rik level,we introduced into primary neuronal cultures a miRNA precursor mimic(‘‘pre-mir-185’’),which is pro-cessed into mature miRNA,or a scramble precursor(‘‘prescram-ble’’)with no homology to the mouse genome,which serves as a control for nonspecific effects of small RNA expression. Twenty-four hours posttransfection,there was a decrease in the levels of2310044H10Rik in pre-mir-185-transfected neurons when compared to prescramble-transfected neurons(p<0.01; Figure3B).In a complementary experiment,introduction of an anti-miR-185LNA oligo or a scramble control oligo resulted in an increase of2310044H10Rik mRNA levels in anti-miR-185-transfected cells when compared to scramble-transfected cells (p<0.05;Figure3C).Taken together,these results confirm that2310044H10Rik expression in primary neurons is under the repressive control of miR-185.Essentially identical results were obtained when2310044H10Rik expression was assayed in N18cells(Figures3D and3E).Therefore,we used this cell line to further characterize the miR-185-mediated inhibition.To test whether the inhibition of miR-185on2310044H10Rik expression is30UTR dependent as predicted by TargetScan and mirDB(see above),2310044H10Rik30UTR-fused luciferase reporter genes were cotransfected with either pre-mir-185mimic or prescramble into N18cells.Although prescramble did not affect the reporter activity,introduction of pre-mir-185mimic led to a dramatic decrease of luciferase activity as compared to the prescramble control(p<0.001for all pre-mir-185concen-trations used;Figure3F).To investigate whether miR-185-medi-ated repression is specific and operates directly via the two binding sites predicted by TargetScan(Figure3A),we engi-neered luciferase reporters carrying mutated versions of 2310044H10Rik30UTR with either individual or both miR-185 binding sites mutated(Mut1:Site1mutant;Mut2:Site2mutant; Mut1&2:Site1and2mutants,see Extended Experimental Procedures).The pre-mir-185mimic significantly reduced the luciferase activity of the WT reporter to$25%relative to a control reporter without30UTR,whereas it reduced the luciferase activ-ities of the Mut1and Mut2reporters to80%(p<0.01)and33%(C and D)Temporal expression of2310044H10Rik(Mirta22)in the PFC(C)and HPC(D)of Df(16)A+/Àand WT littermate mice as monitored by qRT-PCR(n=9–10 for each group).(E)Increased expression of endogenous2310044H10Rik(Mirta22)in DIV9hippocampal neurons isolated from Df(16)A+/Àanimals as assayed by qRT-PCR(n=3 each genotype).Expression levels in mutant neurons were normalized to WT neurons.Results are expressed as mean±SEM.*p<0.05,**p<0.01,and***p<0.001(Student’s t test).See also Figure S2and Tables S1and S2.Cell152,262–275,January17,2013ª2013Elsevier Inc.265(p <0.05),respectively (Figure 3G).Notably,the pre-mir-185mimic could not repress luciferase activity driven from a mutant reporter where both binding sites are simultaneously disrupted (Figure 3G).Thus,both miR-185cognate binding sites have an impact on the 30UTR-mediated regulation of 2310044H10Rik expression,although the site disrupted in the Mut1reporter (Site 1)seems to be the major target site via which miR-185directly exerts its repressive effect.We further addressed the dependence of 2310044H10Rik 30UTR reporter repression on the levels of miR-485or miR-491,which are also predicted to target binding sites in the 30UTR ofthe 2310044H10Rik gene.Both of these miRNAs are modestly downregulated in the HPC of the Df(16)A +/Àmice due to Dgcr8hemizygosity (Figure S3B).The pre-miRNA mimics of either miRNA modestly but significantly reduced the luciferase activity of the 30UTR-fused reporter compared to the prescramble control (pre-mir-485:27%,p <0.05;pre-mir-491:35%,p <0.05;Figure 3H).A three-factor ANOVA indicated that all three miRNAs (miR-185,miR-485,and miR-491)and their inter-actions have significant impact on the luciferase activity with the exception of the interaction between miR-485and miR-491(Table S3).Figure 3.miR-185Directly Targets and Represses 2310044H10Rik(A)Structure of the 30UTR of 2310044H10Rik (Mirta22)showing miRNA binding sites predicted by TargetScan or mirDB.Blocks in mouse 2310044H10Rik (Mirta22)30UTR that are highly conserved in rat and human orthologs are shown below the mouse 30UTR.Evolutionary conservation is also assessed by the ‘‘30-way multiz alignment and conservation analysis’’in the USCS browser,with conserved blocks indicated by green peaks.miR-185and miR-485binding sites located within the conserved blocks are shown in red.(B and C)qRT-PCR quantification of endogenous 2310044H10Rik (Mirta22)in DIV7hippocampal neurons.Expression levels in anti-miR-185-treated and pre-mir-185-treated neurons were normalized to expression levels under respective controls.(B)Increased expression levels of Mirta22in neurons transfected with anti-miR-185at DIV5(n =5,each treatment).(C)Reduced expression levels of Mirta22in DIV9hippocampal neurons transfected with pre-mir-185mimic at DIV7(n =3,each treatment).(D and E)qRT-PCR quantification of endogenous 2310044H10Rik (Mirta22)in N18cells.Expression levels in pre-mir-185-treated and anti-miR-185-treated cells were normalized to expression levels under respective controls.(D)Reduced expression levels of Mirta22in cells transfected with pre-mir-185mimic (n =3,each treatment).(E)Upregulation of Mirta22in cells transfected with an anti-miR-185LNA oligo (n =3,each treatment).(F–H)Repression effects of pre-mir-185,pre-mir-485,and pre-mir-491on Mirta2230UTR were examined by a dual-luciferase reporter assay (see Experimental Procedures ).Values are Renilla luciferase levels relative to firefly luciferase levels and normalized to the relative expression levels under prescramble treatment (F and H)or to the relative expression levels from plasmid with no 30UTR (G)(n =3for each condition).Pre-mir-185significantly decreases the 2310044H10Rik (Mirta22)30UTR reporter expression over a concentration range of 10–0.01nM (F).Pre-mir-185-mediated repression on 2310044H10Rik (Mirta22)30UTR reporter expression depends on conserved miRNA binding sites (G).Pre-mir-485and pre-mir-491significantly decrease the 2310044H10Rik (Mirta22)30UTR reporter expression (H).Results are expressed as mean ±SEM.*p <0.05,**p <0.01,and ***p <0.001(Student’s t test).See also Figure S3and Table S3.266Cell 152,262–275,January 17,2013ª2013Elsevier Inc.Taken together,thesefindings suggest that the persistent elevation of2310044H10Rik levels observed in Df(16)A+/Àmice is likely the result of the combined hemizygosity at miR-185 and Dgcr8loci.Although more than one miRNA contributes, the major effect is due to the dramatic downregulation of miR-185.Consistent with this notion and the less profound reduction of miR-185in Dgcr8+/Àmice(Figure1E),2310044H10Rik is only modestly upregulated in this strain(Figure S3A).Interestingly, a comparison between the30UTR of human and mouse ortho-logs(Figure3A)reveals that miR-185cognate Site1as well as one miR-485binding site are located within a highly conserved region,suggesting that these sites are critical in regulating the levels of the human ortholog(C19orf63).Consistent with this expectation,introduction of pre-mir-185into human293T cells resulted in a significant decrease of endogenous C19orf63levels (Figure S3C).In addition,similar to the pattern observed in the mouse brain,expression of C19orf63decreases in infant brain as depicted in BrainSpan database(). It is noteworthy that inspection of our gene expression data set as well as qRT-PCR analysis of a sample of eight high-likeli-hood miR-185targets identified by more than one prediction program did not reveal any additional significant changes of transcript levels in the brains of Df(16)A+/Àmice(Figure S3D). Furthermore,unlike2310044H10Rik,none of the other top upre-gulated protein-coding genes(shown in Figure2B)are consis-tently altered in both HPC and frontal cortex of E17,P6,and adult Df(16)A+/Àmice,and only one of them(B3gat1,see below)is predicted to contain miR-185seed sites in its30UTR.Overall, although additional downstream targets of miR-185likely exist (see below),our analysis suggests that2310044H10Rik re-presents the major downstream effector of miR-185and a major hub target of miRNA dysregulation due to the22q11.2 microdeletion.Due to confirmed miRNA-mediated regulation, we renamed the gene Mirta22(miR NA ta rget of the22q11.2 microdeletion).Mirta22Encodes a Neuronal Protein Residingin the Golgi ApparatusMirta22encodes a28kDa protein without any known sequence homology or functional domain(/uniprot/ Q3TAS6).The murine ortholog is located on mouse chromo-some7and contains seven coding exons.The human ortholog (C19orf63)is located on chromosome19q13.33and encodes a protein with92.3%identity to the murine protein(Figure4A). One mouse reference sequence(isoform1)is reported in Gen-Bank,whereas two C19orf63isoforms(isoform1and2)are re-ported in GenBank and in the literature(Junes-Gill et al.,2011). The protein encoded by isoform1is predicted to contain a N-terminal signal peptide,as well as a transmembrane segment (Figure4A,red rectangles),which separates a long N-terminal region from a short C-terminal segment that contains a polygly-cine tail with unknown function.Isoform2differs from isoform1 by an alternatively spliced exon located after exon6.The protein encoded by isoform2is shorter by eight amino acids,contains the N-terminal signal peptide but not the transmembrane segment,and is predicted to be secreted(Figure4A).We raised a polyclonal antibody against a segment of the protein that is conserved in the mouse and human orthologs(amino acids 207–226,Figure4A,green rectangle;see Extended Experi-mental Procedures;Figure S4).We validated the specificity of the antibody using a number of assays(see Extended Experi-mental Procedures;Figures S4A–S4C)and showed that it can also detect the secreted form of the protein in293T cell cultures (Figure S4D).Western blot assays of protein extracts from the brain of Df(16)A+/Àmice and WT littermates showed the expected increase(25%)in Mirta22levels in mutant mice(Fig-ure4B).A similar magnitude increase of the Mirta22immunocy-tochemical signal was observed in Df(16)A+/Àcultured neurons, as compared to WT neurons(Figure4C).Immunostaining of neuronal cultures showed that Mirta22is primarily a neuronal protein(Figure4D,top).At the subcellular level,it is found primarily in the soma,where it colocalizes with the Golgi apparatus marker GM130(Nakamura et al.,1995).As the neurons mature,it is also found in vesicles and tubular-like clusters within the dendritic shafts(Figure4D,middle).Mirta22 immunoreactivity was not detected in cultures stained with pre-immune serum(Figure S4E)and was diminished by64%in Mirta22shRNA-transfected neurons(Figures S4F and S4G, lower panel).Immunohistochemical analysis demonstrated that Mirta22is widely distributed in the brain,where it is localized in neurons(Figure4D,bottom).miR-185Reduction Results in Coordinated Mild Dysregulation of Golgi-Related GenesAccumulating evidence suggests that miRNAs may target functionally connected genes,often in a developmental stage-specific manner(Tsang et al.,2010;Zhang et al.,2009).Consis-tent with this notion,functional annotation clustering analysis of2,695out of2,708predicted miR-185targets(TargetScan Mouse v.5.2)included in the DAVID Mus musculus gene functional annotation database() identified as the top enriched gene cluster(gene count=159, Enrichment Score=8.56,FDR-corrected p=2310À9)the Gene Ontology(cellular component)term‘‘Golgi apparatus’’(Figure5A).Gene set enrichment analysis(GSEA)on the2,708 predicted miR-185targets ranked based on the gene expression profile of Df(16)A+/Àmice also indicated that the Gene Ontology terms‘‘Golgi apparatus part’’and‘‘Golgi apparatus’’were among the top20gene sets in the adult HPC(Figure5A).A global perspective on the enrichment of this miR-185target gene set among the differentially expressed genes in the Df(16)A+/Àmice showed a significant enrichment in the adult HPC expres-sion profile(p=5310À4)where,as expected,most of the top genes were upregulated(n=34),and only four genes were down-regulated(p<0.005,Figure5B;Table S4).A considerably more modest enrichment was suggested for the E17(p=0.02)and P6 HPC(p=0.016)profiles(Figure S5).Interestingly,there was no significant enrichment within the PFC profiles in any of the three ages tested(E17:p=0.6311;P6:p=0.1326;adult:p=0.244). Expression changes were modest,with only4of159Golgi-related probe sets included among the top100in the adult HPC. Altered miR-185Levels Contribute to Structural Alterations of Df(16)A+/ÀNeuronsDf(16)A+/Àmice show impaired formation of dendrites and spines in the HPC(Mukai et al.,2008)and the PFC(Figures Cell152,262–275,January17,2013ª2013Elsevier Inc.267S6A–S6C),which are faithfully recapitulated in primary neuronal cultures.Impairment in these processes in Df(16)A +/Àmice could only be partially accounted for by the 50%decrease in the levelsof Dgcr8(Fe´nelon et al.,2011;Stark et al.,2008).Localization of Mirta22within the Golgi apparatus and dendritic shafts suggeststhat diminishment of the miR-185repression on Mirta22levels may also contribute to these deficits.To test this hypothesis,we first asked whether reduction of miR-185levels results in deficits in dendritic and spine develop-ment similar to those observed in Df(16)A +/Àneurons (MukaiFigure 4.Genomic Structure,Neuronal Expression,and Subcellular Localization of 2310044H10Rik(A)Top view shows the structure of mRNA transcripts of 2310044H10Rik (Mirta22)and its human ortholog,C19orf63.RefSeq reports a 2310044H10Rik (Mirta22)transcript with seven exons (blue rectangles),which is predicted to encode a signal peptide and a transmembrane domain (red rectangles).The peptide epitope used to generate a polyclonal antibody is marked by a green rectangle.For C19orf63,RefSeq reports two alternatively spliced transcripts:one that encodes a predicted transmembrane protein and one with an additional exon that encodes a predicted secreted protein.Bottom view shows protein sequence alignment of predicted transmembrane isoforms encoded by 2310044H10Rik (Mirta22)and its human ortholog.Black blocks indicate completely conserved residues,gray blocks indicate similar residues (defined by Boxshade default similarities),and white blocks indicate different residues.(B)Upper view presents representative western blot assays of 2310044H10Rik (Mirta22)in PFC lysates prepared from Df(16)A +/Àanimals and WT littermates.a -tubulin is used as loading control.Lower view is quantification of 2310044H10Rik (Mirta22)protein level in PFC of Df(16)A +/Àand WT animals (n =9each genotype).Expression levels in mutant mice were normalized to WT littermates.Results are expressed as mean ±SEM.**p <0.01(Student’s t test).(C)Quantification of 2310044H10Rik (Mirta22)immunocytochemical signals in Df(16)A +/Àand WT cultured neurons (n =31for Df(16)A +/À;n =34for WT).Expression levels in mutant neurons were normalized to WT neurons.Results are expressed as mean ±SEM.*p <0.05(Student’s t test).(D)Upper panel shows that 2310044H10Rik (Mirta22)colocalizes with neuron-specific marker NeuN,but not with glia-specific marker GFAP,in cultured hippocampal neurons at DIV20.Middle panel shows that 2310044H10Rik (Mirta22)(green)colocalizes with Golgi-specific marker GM130(red)in the soma.2310044H10Rik (Mirta22)is also found in vesicles and tubular-like clusters in the dendrites,which are highlighted by the dendritic marker MAP2(blue).Lower panel presents distribution of 2310044H10Rik (Mirta22)protein in adult mouse brain.Sections were stained with 2310044H10Rik (Mirta22)antibody.Images were taken at 43,103,203,and 403magnifications as indicated.Red boxes in 43,103,and 203images outline the area shown in 103,203,and 403images,respectively.See also Figure S4.268Cell 152,262–275,January 17,2013ª2013Elsevier Inc.。
微阵列图像降噪和自动定位_张琛

吞吐量分析的全自动划格方法, 对微阵列图像进行自动定位, 对于基因表达微阵列图像精度可
达到 97% .
关键词: 自动划格; 图像降噪; 小波变换; 微阵列图像
中图号: T N911. 73
文献标志码: A
随着基因芯片实验技术的不断发展, 数据精 度以及数据处理效率要求越来越高, 而目前常用的 划格方法虽然可以达到合适的精度, 但由于人工的 参与容易带来额外误差, 而且在处理大数据量信息 时效率极低.
F ig . 3 Wavelet denoising imag e
图 4 两种方法结合后的图像 CPSN R= 64. 658 4 dB
F ig . 4 Co mbinat ion of the tw o methods
2 微阵列自动化格
位样点. 为了实现自动的角度调整, 文中提出了一 种功率谱子阵列角度调整算法. 用来确定微阵列图
图 1 背景图像 Fig . 1 Backgr ound imag e
第二种方法是使用小波变换滤除图像中的高 频噪声[ 5] . 小波包分析能够为微阵列图像提供精细 的分析方法, 对图像中高频部分进行细分, 根据被
分析图像的特征, 自适应选择相应频带, 更加精确
的滤除高频噪声. 因为 Sy mletA ( sy mN) 小波族具有较好的对称
n- 1
n- 2
% % 令
Y=
1 n-
1 i= 1 D ( i)
+
1 n-
2
i=
1
T(
i)
( 9)
将 Y 作为阈值, 当 D( i) 值小于Y 时, 则将 D( i )
作为样点 i 和样点i + 1 间距估计, 当差分运算函数
significance analysis of microarrays

基因芯片显著性分析(Significance Analysis of Microarrays,SAM)是一种统计方法,用于在基因表达数据中识别具有显著差异表达的基因。
该方法可以帮助研究人员确定哪些基因在不同实验条件下表达水平发生显著变化,从而揭示与特定生物过程或疾病相关的基因。
SAM方法的基本原理是比较两个或多个实验条件下的基因表达数据,通过计算每个基因的统计量(例如t检验的t值),评估基因表达差异的显著性。
该方法允许研究者设定一个阈值,以确定哪些基因的表达差异是显著的,同时控制假阳性率(即错误地认为基因表达有显著差异的概率)。
与传统的统计方法相比,SAM具有更高的灵敏度和特异性,能够更好地识别真正具有显著差异表达的基因。
此外,SAM还提供了一种可视化工具,称为SAM图(Significance Analysis of Microarrays plot),用于直观地展示基因表达数据的分布和显著差异表达基因的识别结果。
总之,基因芯片显著性分析(SAM)是一种强大的统计方法,可用于识别基因表达数据中的显著差异表达基因。
它在生物医学研究中具有广泛应用,有助于揭示生物过程和疾病的分子机制。
基因芯片技术原理

基因芯片(Gene chip)
Southern & Northern Blot Dot Blot
Macroarray
Microarray
基因芯片的研发历史
八十年代末期俄美科学家提出“杂交法测序”
1992年世界第一块原位合成基因芯片在美国 Affymetrix诞生(1994第一张商业化芯片)
1995年世界第一块微矩阵基因芯片在Stanford大 学实验室诞生
技术背景
随着传统技术的不断改进,基因信 息分析规模不断扩大。 • -人类基因组计划的需求(HGP) • -后基因组时代的需求(功能基因 组学方向)
生物芯片 (Biochips)
将大量生物识别分子按预先设置的排列固定于一种载 体(如硅片、玻片及高聚物载体等)表面,利用生物分子 的特意性亲和反应,如核酸杂交反应,抗原抗体反应等来 分子各种生物分子存在的量的一种技术。
image
Commercial chip
• Human • Rat • Mouse • Drosophila • E. coli • Yeast • Zebrafish
基因芯片的应用
– 特定基因检测 – 突变检测 – 多态性分析 – 基因表达谱
•生物信息学的工具 •基因相关性研究 •基因功能 •药物设计和开发 •潜在反义试剂开发 •个体化医疗 •身份识别 •基因诊断 •其他与生物有关的领域
Removable Tip Orifice
Controller
直接点样法 (stanford, brown)
Spotter head
基因芯片技术流程
基因芯片的设计制备 杂交 检测
数据分析
制备方法及点样仪器
靶基因的标记制备:标记方法 杂交:杂交液、杂交温度、洗涤条件
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Valutazione di software di analisi di microarray basato su simulazioni di immagini da dati realiOverviewIntroduction–Analysis of Microarray images•Addressing or gridding•Segmentation•Intensity extractionMicroarray results: how accurate are they? Evaluation by simulation–Statistical analysis of real data–Simulation Model–Testing–An example of applicationConclusionsReferencesIntroductioncDNA microarray technologies have large diffusion inbiological research field–For studying gene expression in many different organism –To large-scale gene discovery–For polymorphism screening and mapping of genomic DNA clonesFully automated and reliable software analysissystems are required to process the large amount of data producedEffective testing and evaluation of employedcomputational approaches is still an open problemC. C. Xiang, and Y. Chen, “cDNA microarray technology and its applications ”, in Biotechnology Advances, vol. 18, 2000, pp. 35–46.Microarray technologyAnalysis of microarray images A laser scanner detectssample fluorescence–Cy3 probe at 532 nm–Cy5 probe at 633 nmCombined RGB color imagerepresents differentiallyexpressed genes–The fluorescence intensitiesare stored as 16-bit imageswhich we view as “raw”dataImage processing softwareanalyzes red and greenhybridization intensities andred/green ratio for everygene on the array, log2(R/G)Analysis of microarray images (cntd)The digitalization process produces, for each pixel, asignal that represents the total fluorescence in the region corresponding to that pixelWhen properly processed, this signal shouldcorrelate to the area density of dye molecules Addressing or griddingSegmentationIntensity extractionY. H. Yang, M. J. Buckley M, et al., “Comparison of methods for image analysis on cDNA microarray data ”, in J. Comp. Graph. Stat., 11, 2002, 108-136Addressing or griddingIt is the process ofassigning coordinatesto each of the spotsAutomating this part of the procedure permitshigh throughputanalysisAddressing or gridding (cntd.)Separation between rows and columnsIndividual translation of grids–caused by slightvariations in print-tippositionsSeparation between rows and columns ofspotsOverall position of the array in the imageAddressing or gridding (cntd.)Misregistration of the red and greenchannelsRotation of the array in the imageSkew in the arraySegmentationIt allows the classification of pixels either as foreground (i.e. the spot of interest) or as backgroundUsed methodsFixed circle segmentation–ScanAnalyzeAdaptive circle segmentation–GenePixAdaptive shape segmentation(watershed, seeded region growing SRG)–SpotHistogram segmentation (Otsu)–QuantArraySegmentation (cntd.)In microarray image analysis, we are inthe rather unusual situation where thenumber of features (spots) is knownexactly a priorithe approximate locations of the spotcenters are determined at theaddressing stageQ. Li, C. Fraley, R. E. Bumgarner, K. Y. Yeung, and A.E. Raftery, “Donuts, scratches and blanks: robustmodel-based segmentation of microarray images ”,in Bioinformatics, 21:12, 2005, pp. 2875–2882Intensity extractionThis step includes calculating for each spot on the array–red and green foreground fluorescence intensity pairs (R,G)–background intensities•Measured fluorescence intensity includes a contribution which is not specifically due to the hybridization process (background correction)–quality measures•Measures of–Spot size–Spot shape–Background intensity vs foreground intensityIntensity extraction (cntd.)Most microarray analysispackages define theforeground intensity as themean or median of pixelvalues within the segmentedspot maskMore variation exists in thechoice of backgroundcalculation method–Example: taking the medianof values in selected regionssurrounding the spot maskSpot maskQuantArrayScanAnalyzeSpot, GenePixMicroarray results: how accurate are they?Several inconsistencies from different commerciallyavailable systems–Inconsistent sequence fidelity of the spotted microarrays –Variability of differential expression–Low specificity of cDNA microarray probes–…Conclusions: In view of the pitfalls, data frommicroarray analysis need to be interpreted cautiously J. Quackenbush, “Computational analysis of microarray data ”, in Nat. Rev. Genet., vol. 2, 2001,418-427.R. Kothapalli, S. J Yoder, S. Mane, and T. P. Loughran Jr, “Microarray results: how accurate are they?”, in BMC Bioinformatics, 3:22, 2002Modeling microarray experimentsA microarray simulation model can be used tovalidate different kinds of data analysis algorithms –Obtaining realistic biological measurement data–Referring to valid ground truth dataThe simplest approach to generate the ground truthdata is to sample data randomly from a specific distribution.–the distribution and its parameters can be estimated from real measurements.–the ground truth data can be obtained by sampling a simulated ideal distribution with estimated parametersM. Nykter, T. Aho,M. Ahdesmäki, P. Ruusuvuori, A. Lehmussola, and O. Yli-Harja, “Simulation of microarray data with realistic characteristics ”, in BMC Bionformatics, 7:349, 2006Errors and noisesSlide manufacturing–subarray drifting from idealrectangular layout.Slide hybridization–Spot blending–Scratches–Air bubbles–Background noiseSlide scanning–Spot saturation–Channels misalignment–Translation, rotation, skewY. Balagurunathan, E. R. Dougherty, Y. Chen, and M. L. Bittner, J. M. Trent, “Simulation of cDNA microarrays via a parameterized random signal model ”, in Journal Biomed Opt, 7(3), 2002, pp. 507-523Simulation of cDNA microarraysOur approachTo provide researchers with a tool for testing and evaluating theperformance of analysis softwareCharacteristics of a given microarray experiment are capturedfrom public databases–Extracted data are then used for generating a synthetic microarray image and the corresponding ground truth data (i.e. the gene expression values).–Given a particular experiment, and/or a specific hardware, and/or a software tool for microarray image analysis and so on, we use raw data obtained in identical or similar conditions to model simulated images with known values of gene activation.Using the simulated images as benchmark, one can estimateexpected errors and choose the most suitable analysis software for the real experiment.Statistical analysis of real dataIn order to simulate realistic microarray images,public databases could be used to chooseparameters of the model of spot and grid generation –Stanford MicroArray Database•/Grid geometry, spot locations, and other details arerecovered from results file (gpr format)I. Infantino, C. Lodato, S. Lopes, “Testing and evaluation of microarray image analysis software ”, 2nd Intl. Conf. on Complex, Intelligent and Software Intensive Systems (CISIS 2008), Intl. Workshop on Intelligent Informatics in Biology and Medicine (IIBM 2008), Barcelona, Spain, March 4th –7th, 2008Spot model: feature positionsExperiment #28370 in Stanford Microarray Database–Related to the normal tissue of hyperinsulinemic clamp in humanmuscle in diabetes–Tiff images have dimension 5556 x 1952–43200 spots–Results saved as gpr fileTo transform the coordinate system from nm to pixels–image origin O 1=(x 01,y 01) is (920,7720)–pixel size S pixel is 10 nm–coordinates (x f , y f ) and diameter diaf in pixels of features areobtained as:x f = x-x 01 / s pixely f = y-y 01 / s pixeldia f = dia / s pixelSpot model: background and foregroundBackground and spot pixelsof considered area areseparated by K-meansalgorithm–looking for two intensitiesclusters grouping bysquared Euclidean distance–K-means initial seed pointsare the first pixel of theregion (upper-left corner)and the central oneHoles are found by labelingconnected regions using 4-connectionSpot model: feature intensitiesmedian of spot pixel intensities medians–MM spot_ch1, MM spot_ch2mean of spot pixel intensities variances–Var spot_ch1, Var spot_ch2mean of correlation of spot pixel intensities between channels–Corr spot_ch1, Corr spot_ch2variance of correlation of spot pixel intensities between channels–VarCorr spot_ch1, VarCorr spot_ch2Noise and defectsBackground noisemedian of background pixel intensities medians–MM b_ch1, MM b_ch2mean of background pixel intensities variances–Var b_ch1, Var b_ch2Spot alignmentmean of x misalignment between barycentre of spot and centre of sub-region which includes it–M x_mis_ch1, M x_mis_ch2mean and variance of y misalignment–M y_mis_ch1, M y_mis_ch2Noise and defects (cntd.)Holes within spotmean of number of holes–M n_hole_ch1, M n_hole_ch2mean of distances of holes from spot barycentre–M d_hole_ch1, and M d_hole_ch2ValuesSimulation Modelspots generally have a non-regular shape–some morphological distortion instead of being perfectly circular–spots with doughnut shape are frequently observed in real microarray imagesShape can be effectively modeled by a linearcombination of two bivariate Gaussian distributions –Given a bivariate Gaussian distribution G(x,y)–and a set of n secant planes {P n (x ,y )}–each surface can be expressed by the last element of a sequence {C n (x ,y )} of linear combination between functions describing a surface and the corresponding secant planeSimulation Model (cntd.)denoting with m the generic surface of the envelope, the model can be expressed by the followingequations:Cm0(x,y)=Gm(x,y)Cmn (x,y)=Cmn-1(x,y)for each x,y in D|Cmn-1(x,y)≤Pmn(x,y) (eq. 1)Cmn (x,y)=amnCmn-1(x,y)+bmnPmn(x,y)for each x,y in D|Cmn-1(x,y)>Pmn(x,y),with amn ,bmnin {-1,0,1,2}(eq. 2)where m=1,2,…M (M no. envelope surfaces),n=1,2,…Nm (Nmno. section planes of m-th surface).Spot shapesThe values of coefficients a mn and b mn, affect the specific type of shape: convex, plane, concaveEquations (1) and (2) describe how the surface is modified by the application of the correspondingsecant planethe surface is generally cut in two regions–The one under the secant plane remain unchanged–whereas the other is modified in accord to the equation (2) There are three different combination modes allowed, namely–clip amn = 0, bmn= 1–dig amn = -1, bmn= 2–lift amn = 2, bmn= -1Spot shapes (cntd.)The previous described procedure is repeated until all the secant planes have been applied, thus producing a single element of the envelopeThe succeeding elements will be defined repeating the same procedure as many times as many are the number of specified GaussianHence, the final surface is obtained enveloping all the single surfaces:S(x,y)=max(C m N m (x,y))The resulting function S(x,y) represents the spatial distribution of the simulated spot brightness.Shape parametersThe parameterscharacterizing eachGaussian distribution are–the standard deviationalong x ,y -direction (σx ,σy )–the correlation factor r–the peak coordinates(peak x , peak y )Three coefficients (a , b , c )are needed in order tospecify each correlated secant plane.Simulation ParametersSimulation ParametersTestingSimulated microarray image #28370 generated has beenprocessed by three different software tools:–one based on seeded region growing algorithm (SRG)–one based on Otsu algorithm–one based on adaptive circle segmentation (ACS)ConclusionsThe proposed methodology allows to capturespecificities of a given microarray experiment and to create simulated images to evaluate errors of analysis software toolsThe proposed model is simple and includes onlyparameters that can be determined in fast and robust way starting from real dataOur idea is that does not exist a segmentation algorithm or software definitely better than all the others, but it is possible to choose the one assuring the best precision for given experiment andtechnology.。