第二部分+Chem3D
Chem3D讲义22

异戊二烯3D模型可输入“CH2C(CH3)CHCH2”。
4-甲基-2-戊醇 3D模型可输入 “CH3CH(CH3)CH2CH(OH)CH3”。
三、Chem3D的使用
3、3D模型的建立方法
(3)利用子结构(Substructure)建立模型 Chem3D提供了子结构库,用户可以选 择其中的子结构,然后将它们拼装起来, 形成复杂结构。 操作: [View]—>[Parameter Tables]—>[Substructure], 选定所需要的子结构后, 右键“Copy”, 然后在菜单栏上按 “Paste”,获得3D模型
• 编辑快捷键与其他软件如Word相同,常用的与图形编辑有关的快捷键为: • Ctrl + A:全选 • Ctrl + C:复制 • Ctrl + V:粘贴 • Ctrl + X:剪切
• F9:字符下标
• F10:字符上标 • Ctrl + R:旋转图形,可以设定旋转角度。 • Ctrl + K:改变图形大小,可以设定键长。 • Ctrl + Shift + H:水平翻转图形。 • Ctrl + Shift + V:垂直翻转图形。
13C核磁共振图谱
1H核磁共振图谱
分析结构估计性质
• 执行【View】/【Show Analysis Window】菜单命令, 弹出如图所示的分析窗口。 • 执行【View】/【Show Chemical Properties Window】 菜单命令,弹出如图所示的 化学性质窗口
元素周期表
3D模型区
平 面 图 作 图 区
数据消息窗口
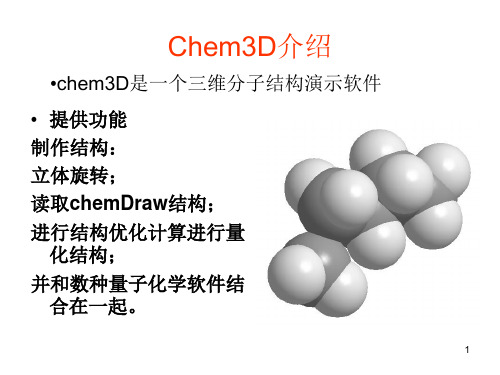
二、Chem3D介绍
平面作图区: 与ChemDraw基本一样 虚键 缩放
Chem3D 使用指南说明书

Molecular MechanicsMostly From "A Guide to Using Chem3D"Molecular mechanics is a non-quantum mechanical technique for calculating energies and some properties of molecules. Because of the way it treats the nuclei and electrons in the molecule, it is a comparatively fast computational method.Molecular Mechanics Theory in BriefMolecular mechanics describes the potential energy of a molecule in terms of a set of potential energy equations that are reminiscent of classical mechanics in physics. The potential energy equations to calculate the energy, the “atom types” that define different atoms in a molecule, and the parameters/constants used in the equations are known as a force-field.Molecular mechanical methods are based on the following principles:Nuclei and electrons are lumped together and treated as unified atom-like particlestypically treated as spheres.Bonds between particles are viewed as harmonic oscillators.Individual potential functions are used to describe the different interactions: bondstretching, angle bending, and torsional (bond twisting) energies, and through-space(non-bonded) interactions.Potential energy functions rely on empirically derived parameters (force constants,equilibrium values) that describe the interactions between sets of atoms as found foratoms in their “natural” or “ideal” state.The sum of interactions for a particular spatial distribution (conformation) of atoms is a measure of intramolecular strain that is relative to a hypothetical situation.Molecular mechanical energies have no meaning as absolute quantities. They can only be used to compare relative steric energy (strain) between two or more conformations of the same molecule or for stereoisomers or for other molecules whose energies are calculated using identical parameters.The total potential energy of a molecular system is the sum of individual potential components. Simple molecular mechanics force fields include bond stretching, angle bending, torsion, and van der Waals interactions in their make-up. The sums extend over all bonds, bond angles, torsion angles, and non-bonded interactions in the molecule. Modern force fields add other termsfor greater accuracy. These properties are easiest to describe mathematically when atoms are considered as spheres of characteristic radii....VDW torsion bend stretch V V V V VTorsionBond Stretch Non-bonded interactionsMolecular mechanics typically treats atoms as spheres, and bonds as springs. The mathematics of spring deformation (Hooke's Law, F = -kx ) is used to describe the ability of bonds to stretch, bend, and twist. The potential energy, V, of a deformation is221212/1)( )(kx dx kx dxxFV x x xxThe simplest expressions for some of the individual potentials are shown below. Additional constants are added to these equations to refine the parameterization.Bond Stretching : bond stretching between directly bonded atoms (see previous example of Hooke’s Law).20)(2/1l l k V s stretchk s is the stretching force constant that is determined empirically. l is the actual bond length in the molecule and l 0 is the “natural” bond length. For example, if the strain-free bond length of a Csp3-H bond is 1.10Å, then any deviation (l-l 0), either longer or shorter, will increase the energy of the molecule. The bond-stretching term is summed over all bonds in the molecule.Angle Bending : angle bending between atoms that are geminal to each other (bonded to the same central atom).20)(2/1b bending k VK b is the bending force constant that is determined empirically. is the actual bond angle in themolecule and 0 is the “natural” bond angle. If the optimal bond angle for H-Csp 2-Csp 3 is 122°, then any change in angle (θ-θ0), either wider or narrower, will increase the energy of the molecule. The angle-bending term is summed over all bond angles in the molecule.Torsion Energy : torsional angle rotation between atoms that are vicinal (bonded to adjacent atoms) to each other.) cos 1(2/10n V V torsionHere, V 0 is the barrier to free rotation for the “natural” bond , n is the periodicity of the rotation (number of cycles in 360°), and ω is the tors ion angle. The torsion term is summed over all rotating bonds in the molecule.Non-bonded interactions : atoms (greater than two bonds apart) interact through van der Waals attraction, steric repulsion, and electrostatic attraction/repulsion depending on their distance from each other. For two approaching non-bonded atoms, the interaction is attractive (London dispersion force) until the atoms get too close and start to repel each other (van der Waals repulsion/steric strain)Dipole-Dipole interactions : bond dipole moments can be used to represent electrostatic interactions in the molecule.0.511.522.5090180270360V a l u e Angle w CosineThe reliability of a molecular mechanical force-field depends on the parameters and the potential energy functions used to describe the total energy of a model. Parameters must be optimized for a particular set of potential energy functions, and thus are not easily transferable to other force fields. Currently, force fields in inorganic and organometallic chemistry are not adequately formulated.The sum of all these terms is called the steric energy of a molecule. Es is only a measure of intramolecular strain relative to a hypothetical situation. By itself, Es has no physical meaning. Because the force fields are parameterized from known bond energies, different parameters are used to calculate the strain energies of structural isomers. Strain energy comparisons are valid only for stereoisomers and conformational isomers. The molecules must have the same connectivity. Some examples of these isomers are anti and gauche butane, axial and equatorial methylcyclohexane, cis and trans 2-butene.。
Chem3D常用功能使用教程
• 模型的输入和输出:以NaCl晶体为例:
• 【File】【Template】选择【NaCl Crystal.C3T】 选择以【Space filling】显示模型。可保存多种格 式,默认格式.c3t。可存为jpg等。
隐藏或显示其它原子
居中 移动到 色彩调节 显示元素符号 显示元素编号 显示实心球 显示点阵表面
调整键长 破坏键 调整键级 就近结合 添加质心 颠倒 调整矩阵
隐藏所选部分 隐藏非选部分 显示所有原子 显示邻近原子 显示背面 定义基团
利用【键】工具建立的结构,键 角及键长可能不正常,应【整理 结构】
选择HOMO 轨道
轨道选项 HOMO和 LUMO各6个
实心显示 (丝网显示 圆点显示 半透明显示)
HOMO轨道 显示
乙烯分子的HOMO轨道
选择LUMO 轨道
LUMO轨道 显示
乙烯分子的LUMO轨道
其他计算
从头算程序:Gamess Gaussian
需另外安装这两个程序。 半经验或分子力学计算
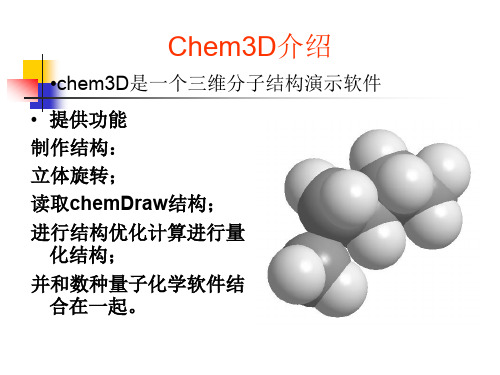
Chem3D介绍
•chem3D是一个三维分子结构演示软件
• 提供功能 制作结构: 立体旋转; 读取chemDraw结构; 进行结构优化计算进行量
化结构; 并和数种量子化学软件结
合在一起。
Chem3D起始界面
主界面
Chem3D起始界面
工具栏
模型类型
三维视图
ChemDraw切换
图标工具栏
模型窗口
扩大(缩小)窗口 状态栏
建立十肽菌素模型
建立十二丙氨酸醇分子模型
chemical3D教程

Chapter 1 Introduction1.1启动Chem3D在开始(Start)菜单中点击所有程序(P), 选择弹出菜单中ChemOffice2004中的Chem3D Ultra 8.0即可启动程序1.2Chem3D 界面结构Chem3D软件的最大特点就是界面结构简单。
和ChemDraw类似,Chem3D 打开后的窗口包含:工作窗口, 信息窗口, 列表窗口, 工具栏, 菜单栏等, 如图1所示。
1.3图形工具栏图形工具栏包含所有能够在工作窗口绘制结构图形的工具,选择这些工具后,光标将随之改变成相应的工具形状。
工具板见图1-2其它的类似于ChemDraw,有兴趣的同学可以参考FTP所提供的英文参考资料Chapter 2 建模简要教程2.1 使用键工具建模首先打开工作窗口(File菜单– New Model)2.1.1 建模在工具栏选择单键工具将鼠标移动至工作窗口,按住鼠标左键拖动鼠标即可绘制简单的乙烷分子。
(注意:View-Settings-Model Build – Rectify 选择上后会自动为所绘制的分子结构加上氢原子)2.1.2 旋转模型:选择旋转工具可以在任意方向选择所绘制的分子。
1:将鼠标移动至工作窗口,按住鼠标左键2:在任意方向拖动鼠标可以旋转模型注意:当拖动鼠标时,会出现一个圆。
在圆内拖动鼠标使得模型绕X和Y轴旋转。
当鼠标在圆外时,模型绕Z轴旋转。
如图所示2.1.3 查看模型分子信息选择选择工具,将鼠标移动至相应原子位置将显示相应的原子序数,元素标识及原子类型。
如下图所示将鼠标移至C-C键上将显示键长及相应的键级在选择原子的同时,按住Shift键可以同时选择多原子,可以通过这样查看相应的键角和二面角。
1:选择C(1),C(2)和H(7)2:将鼠标移至任意的所选择的原子或键上将显示所选择的键角类似的操作可以用来显示二面角。
2.2 修改模型分子2.2.1 修改键类型1:选择双键工具2:按住鼠标左键,从C(1)的位置拖动鼠标至C(2)的位置将乙烷分子更改为乙烯分子。
Chem3D使用说明

Chem3D Practice and Discussion 2
Please draw H2O, NH3 and CH4 and minimize these three molecules. Are their bond angles reasonable? Please draw PF3, PCl3 , PBr3 and PI3 and minimize these three molecules. Are their bond angles reasonable?
Energy
A B
C
Molecular Geometry
Butane不同的穩定分子型態
0º
120º
60º 180º
能量最佳化(Minimization)
選按「MM2」「Minimize Energy」 選按「Display Every Iteration」 於Minimum RMS Gradients 內 輸入「0.01」 選按「Properties」及「Steric Energy Summary」 選按「Run」 想要檢視計算結果必須按一下 ►,如此計算結果才會出現另 一視窗
What does every energy term mean?
以不同的數學方程式描述分子的作用力
C C Bond Stretching = Kb * ( L - L0 ) Angle Bending = Ka * ( A - A 0 )
2 2
Bond Rotation = Kr * cos( n*(T-T0) )
一非常複雜的偏微分方程式
建立分子PF3
選按 ,將滑鼠移至任何位置 按拖曳成一直線後,放開滑鼠 即成C2H6。 選按 選按任一碳原子,請連續按兩次 滑鼠左鍵;於分子模型上方的空 白視窗填寫「P」即磷原子。 選按另外一碳原子,請連續按兩 次滑鼠左鍵;於分子模型上方的 空白視窗填寫「F」即氟原子。 繼續改變其他兩個氫原子。
Chem3D常用功能使用教程 ppt课件
2020/12/2
78
2020/12/2
79
2020/12/2
80
2020/12/2
81
2020/12/2
82
2020/12/2
83
(4) 检查结构能量
• 模型的输入和输出:以NaCl晶体为例:
• 【File】【Template】选择【NaCl Crystal.C3T】 选择以【Space filling】显示模型。可保存多种格 式,默认格式.c3t。可存为jpg等。
• 还有动画显示功能,可让其绕x轴、y轴、z轴转动, 这样可以从不同角度显示结构,更具立体感。 【Analyze】【Spin About Y Axis】,可通过左下 角调整框放大、缩小、播放、暂停、录制等,还 可存成 Windows AVI Movie(*.avi)格式。
2020/12/2
91
(5) 分子轨道(HOMO及LUMO)
首先用 双键 画出乙 烯分子
2020/12/2
92
2020/12/2
显示轨道时 必须选择
93
选择分子轨道
2020/12/2
94
能量随所选轨 道不同而不同
选择HOMO 轨道
2020/12/2
轨道选项 HOMO和 LUMO各6个
实心显示 (丝网显示 圆点显示 半透明显示)
2020/12/2
14
带状模型常用于生物分子
2020/12/2
15
代表用红蓝两种色彩的立体显示
代表用深色显示模型,就是对于模型的部分 按观察者的角度不同着色 代表显示两个立体结构完全相同的模型
Chem3D常用功能使用教程.ppt
在模型窗口沿X、Y轴旋转
19
Z轴旋转指示
选择【轨迹球】工具并按“Alt”键, 使分子模型在模型窗口沿Z轴旋转
20
光标位于原子上, 自动显示原子信息
模型结构信息
21
工具栏显示原子的符号和标号
22
光标位于键上, 自动显示键信息
模型结构信息
23
模型的进一步信息
显示键长变化 显示键角变化
显示二面角 显示所有没有 相邻的原子的
选择以【Space filling】显示模型。可保存多种格 式,默认格式.c3t。可存为jpg等。 • 还有动画显示功能,可让其绕x轴、y轴、z轴转动, 这样可以从不同角度显示结构,更具立体感。 【Analyze】【Spin About Y Axis】,可通过左下 角调整框放大、缩小、播放、暂停、录制等,还 可存成 Windows AVI Movie(*.avi)格式。
ChemDraw中 画出的分子结构
复制粘贴到Chem3D中 63
Chem3D中 画出的分子模型
复制粘贴到 ChemDraw中
64
计算功能演示
(1)查找原子的范德瓦尔斯半径 • 【View】【Atom Types.TBL】窗口,
找到各种元素在不同环境的半径
65
66
67
(2)观察分子的大小 • 建立苯模型。执行【View】【Connolly
88
显示轨道时 必须选择
89
选择分子轨道
90
能量随所选轨 道不同而不同
选择HOMO 轨道
轨道选项 HOMO和 LUMO各6个
实心显示 (丝网显示 圆点显示 半透明显示)
HOMO轨道 显示
91
乙烯分子的HOMO轨道
Chem3D常用功能使用教程.ppt
24
模型的键长数据
25
模型的键长和键角数据
26
乙烯模型中的双键信息
27
键级改动
28
显示氢及孤对电子
29
不显示氢及孤对电子的环己烷
原子名称及序数 30
选择工具,双击原子, 按顺序改变原子序号
31
居中 移动到 色彩调节 显示元素符号 显示元素编号 显示实心球 显示点阵表面
调整键长 破坏键 调整键级 就近结合 添加质心 颠倒 调整矩阵
选择以【Space filling】显示模型。可保存多种格 式,默认格式.c3t。可存为jpg等。 • 还有动画显示功能,可让其绕x轴、y轴、z轴转动, 这样可以从不同角度显示结构,更具立体感。 【Analyze】【Spin About Y Axis】,可通过左下 角调整框放大、缩小、播放、暂停、录制等,还 可存成 Windows AVI Movie(*.avi)格式。
View/Setting/Model Dispaly 菜单或F6键:五种 Space Filling
Wire Frame Sticks Ball-Sticks Cylindrical Bonds
10
带状模型常用于生物分子
11
代表用红蓝两种色彩的立体显示
代表用深色显示模型,就是对于模型的部分 按观察者的角度不同着色 代表显示两个立体结构完全相同的模型
103
104
放大、 缩小
播放、暂 停、录制
滑动块
105
106
Molecular】弹出Suface对话框;【solid】 选项可以选择分子表面的类型; 【Resolution】滑动到右边,其值为100
68
69
70
71
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
102
• 模型的输入和输出:以NaCl晶体为例: • 【File】【Template】选择【NaCl Crystal.C3T】 选择以【Space filling】显示模型。可保存多种格 .c3t jpg 式,默认格式.c3t。可存为jpg等。 • 还有动画显示功能,可让其绕x轴、y轴、z轴转动, 这样可以从不同角度显示结构,更具立体感。 【Analyze】【Spin About Y Axis】,可通过左下 角调整框放大、缩小、播放、暂停、录制等,还 可存成 Windows AVI Movie(*.avi)格式。
96
97
98
99
100
101
其他计算
从头算程序: 从头算程序:Gamess Gaussian 需另外安装这两个程序。 需另外安装这两个程序。 半经验或分子力学计算 Mechanics Mopac (Am1, Mindo/3, Mndo, PM3) 计算方式:计算性质,最小能,过渡态优化, 计算方式:计算性质,最小能,过渡态优化,光谱分析
选择【单键】工具,在模型窗口向右按动
圆柱键模型
14
线状模型
15
棒状模型
16
球棍模型
17
比例模型
18
X、Y轴旋转指示
选择【轨迹球】工具,使分子模型 在模型窗口沿X、Y轴旋转
19
Z轴旋转指示
选择【轨迹球】工具并按“Alt”键, 使分子模型在模型窗口沿Z轴旋转
20
光标位于原子上, 自动显示原子信息
74
75
76
77
78
79
(4) 检查结构能量
80
模型旋转
对位交叉
81
总能量计算
82
计算结果
空间位阻能:0.977kcal/mol
83
模型的二面角数据
84
数据与模型对应
85
显示原子符号及序号
86
按动旋转
数据相应改变
87
(5) 分子轨道(HOMO及LUMO) 首先用 双键 画出乙 烯分子
பைடு நூலகம்103
104
放大、 缩小
播放、暂 停、录制 滑动块
105
106
42
43
整理结构
44
建立Ibuprofen(布洛芬)模型
CH3 H3C CH CH2
COOH CH CH3
45
46
建立十肽菌素模型
47
48
建立十二丙氨酸醇分子模型
49
50
改变显示方式
模型类型窗口:
51
设为线状模型
52
53
设为带状模型
54
α-螺旋模型
55
3. 利用子结构建立模型
68
69
70
71
72
View/Solvent Accessible 菜单 甲苯结构
73
(3)计算分子体积 • 建立苯模型。执行【Analyze】 【Compute Properties】在【Available Properties】选框中双击【Connolly Solvent-Excluded Volume SVEChemPropStd】选项,使之加入到 【Selected Properties】框中。单击ok,
模型结构信息
21
工具栏显示原子的符号和标号
22
光标位于键上, 自动显示键信息
模型结构信息
23
显示键长变化 显示键角变化 显示二面角 显示所有没有 相邻的原子的 距离
模型的进一步信息
24
模型的键长数据
25
模型的键长和键角数据
26
乙烯模型中的双键信息
27
键级改动
28
显示氢及孤对电子
29
不显示氢及孤对电子的环己烷
88
显示轨道时 必须选择
89
选择分子轨道
90
轨道选项 HOMO和 LUMO各6个 能量随所选轨 道不同而不同 实心显示 (丝网显示 圆点显示 半透明显示)
选择HOMO 轨道
HOMO轨道 显示
91
乙烯分子的HOMO轨道
92
LUMO轨道 显示
选择LUMO 轨道
93
乙烯分子的LUMO轨道
94
95
线状模型 棒状模型 球棍模型 圆柱键模型 比例模型
8
带状模型
9
结构不同显示方式
View/Setting/Model Dispaly 菜单或 键:五种 菜单或F6键
Space Filling
Wire Frame
Sticks
Ball-Sticks Cylindrical Bonds
10
带状模型常用于生物分子
32
利用【键】工具建立的结构,键 角及键长可能不正常,应【整理 结构】
33
环己烷的椅式构象
34
2. 利用文本工具建立模型
使用【文本】工具输入结构式
35
按【Enter】自动转化为模型
36
括号表示支链
37
38
整理结构
39
简单优化
观察分子变化
40
4-甲基-2-戊醇模型
41
建立1,2-双甲基环戊烷模型
使用子结构建立模型
56
子结构模型库
57
选择所需的子结构,然后复制
58
粘贴复制结果
59
在子结构上进行编辑
60
4. 使用模板建立模型
61
62
5. ChemDraw和Chem3D信息转换
ChemDraw中 画出的分子结构 复制粘贴到Chem3D中
63
Chem3D中 画出的分子模型
复制粘贴到 ChemDraw中
64
计算功能演示
(1)查找原子的范德瓦尔斯半径 • 【View】【Atom Types.TBL】窗口, 找到各种元素在不同环境的半径
65
66
67
(2)观察分子的大小 • 建立苯模型。执行【View】【Connolly Molecular】弹出Suface对话框;【solid】 选项可以选择分子表面的类型; 【Resolution】滑动到右边,其值为100
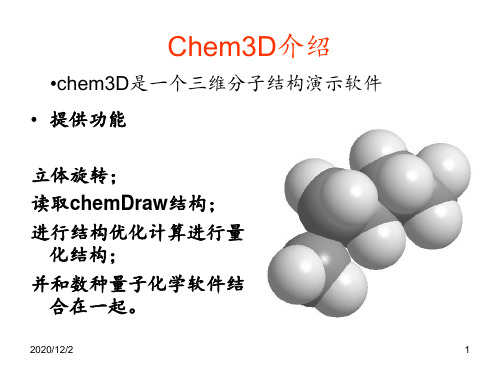
Chem3D介绍
•chem3D是一个三维分子结构演示软件 • 提供功能 制作结构: 制作结构: 立体旋转; 立体旋转; 读取chemDraw结构; 结构; 读取 结构 进行结构优化计算进行量 化结构; 化结构; 并和数种量子化学软件结 合在一起。 合在一起。
1
Chem3D起始界面
2
主界面
Chem3D起始界面
11
代表用红蓝两种色彩的立体显示 代表用深色显示模型,就是对于模型的部分 按观察者的角度不同着色 代表显示两个立体结构完全相同的模型 代表模拟现实的角度观察模型,使模型更 接近真实 代表显示模型的时候将后面的部分以阴影 形式表示,这样模型的立体感更强。
12
13
建立3D模型 1. 利用键工具建立模型
3
工具栏
模型类型
三维视图
ChemDraw切换
模型窗口 图标工具栏
模型数据窗口 扩大(缩小)窗口
消息窗口 状态栏
4
选择 轨迹球 大小调整 单键 双键 三键 虚键 文本 橡皮
5
显示设置
6
弹出信息 立体显示 视频设置 原子标记 模型显示设置 原子显示 结构显示 配体显示
色彩选项
7
显示设置属性
Model Type
原子名称及序数
30
选择工具,双击原子, 按顺序改变原子序号
31
居中 移动到 色彩调节 显示元素符号 显示元素编号 显示实心球 显示点阵表面 调整键长 破坏键 调整键级 就近结合 添加质心 颠倒 调整矩阵 隐藏所选部分 隐藏非选部分 显示所有原子 显示邻近原子 显示背面 定义基团
隐 藏 或 显 示 其 它 原 子