达泊西汀的合成进展_张飞皇
一种达泊西汀中间体的生物合成方法及其中间体[发明专利]
![一种达泊西汀中间体的生物合成方法及其中间体[发明专利]](https://img.taocdn.com/s3/m/cfba04c7b4daa58da1114a84.png)
专利名称:一种达泊西汀中间体的生物合成方法及其中间体专利类型:发明专利
发明人:黄燕鸽,游庆红,袁君,张世忠,许莹,李进,徐纬川
申请号:CN201910307976.6
申请日:20190417
公开号:CN110078632A
公开日:
20190802
专利内容由知识产权出版社提供
摘要:本发明公开了一种达泊西汀中间体的生物合成方法及其中间体,该方法由化合物(2)和化合物(3)作为起始原料,经相转移催化取代反应制得化合物(4),化合物(4)经生物酶转化反应最终制得达泊西汀中间体化合物(1),其反应式如下所示:;本发明的达泊西汀中间体的生物合成方法具有合成路线新颖,简单易行,成本较低,合成产率高,收率高,产品纯度好、产品质量较好、原料廉价易得以及适合于工业化生产等优点,同时所合成的达泊西汀中间体为达泊西汀制备提供了新的中间体原料。
申请人:淮阴工学院
地址:223003 江苏省淮安市洪泽区东七街三号高新技术产业园A12-2(淮阴工学院技术转移中心洪泽分中心)
国籍:CN
代理机构:南京苏高专利商标事务所(普通合伙)
代理人:孙斌
更多信息请下载全文后查看。
盐酸达泊西汀生产工艺

盐酸达泊西汀生产工艺盐酸达泊西汀是一种常用的化学药品,它在医药领域有着广泛的应用。
本文将介绍盐酸达泊西汀的生产工艺。
盐酸达泊西汀的生产工艺主要分为以下几个步骤。
首先,将原料达泊西汀与盐酸溶液进行反应。
这一步骤的目的是将达泊西汀与盐酸结合,形成盐酸达泊西汀。
反应过程中需要注意控制温度和反应时间,以确保反应的完全性和产物的纯度。
接下来,对反应混合物进行过滤和干燥处理。
通过过滤,可以去除反应中产生的杂质和固体物质。
干燥处理可以将产物中的水分去除,提高产物的纯度和稳定性。
然后,对干燥后的产物进行结晶处理。
结晶过程中,可以通过控制温度和溶剂的浓度来促使产物结晶。
结晶后的产物通常具有较高的纯度和良好的结晶形态。
对结晶后的产物进行干燥和粉碎处理。
干燥可以进一步去除残留的水分,提高产物的稳定性和保存期限。
粉碎处理可以将产物研磨成所需的粒度,以满足不同的应用需求。
盐酸达泊西汀的生产工艺中,需要注意的一些关键技术点。
首先,反应温度和反应时间的控制十分重要,这可以确保反应的完全性和产物的纯度。
其次,过滤和干燥处理需要采用适当的方法和设备,以提高产物的纯度和稳定性。
此外,结晶过程中的温度和溶剂浓度的选择也对产物的纯度和结晶形态有着重要影响。
盐酸达泊西汀的生产工艺需要严格遵守相关的安全操作规程。
在操作过程中,应注意防止产生有害气体和溶液的泄漏。
同时,应配备必要的防护设施,如安全眼镜、手套和防护服等,以确保操作人员的安全。
盐酸达泊西汀的生产工艺是一个复杂而严谨的过程。
通过控制反应条件、过滤干燥和结晶处理等关键步骤,可以获得高纯度和良好结晶形态的盐酸达泊西汀产物。
在实际生产中,应严格遵守相关的操作规程和安全要求,确保产品质量和操作人员的安全。
达泊西汀(dapoxetine)路线
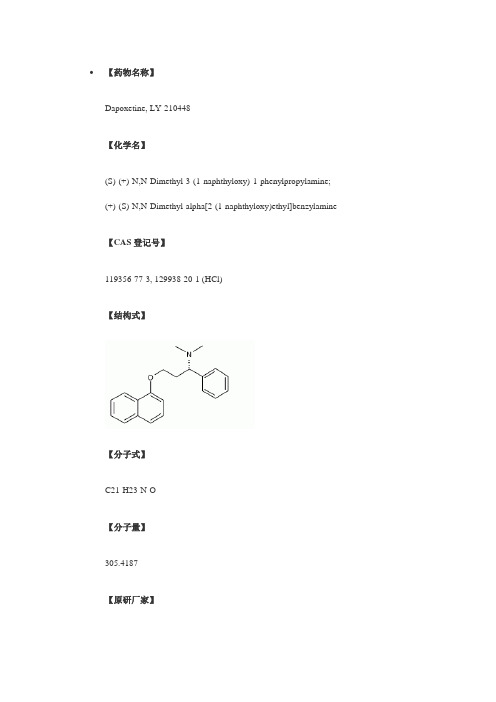
【药物名称】Dapoxetine, LY-210448【化学名】(S)-(+)-N,N-Dimethyl-3-(1-naphthyloxy)-1-phenylpropylamine;(+)-(S)-N,N-Dimethyl-alpha[2-(1-naphthyloxy)ethyl]benzylamine 【CAS登记号】119356-77-3, 129938-20-1 (HCl)【结构式】【分子式】C21-H23-N-O【分子量】305.4187【原研厂家】Lilly (Originator), Dynogen Pharmaceuticals (Not Determined), Alza (Licensee), PPD GenuPro (Licensee)【作用类别】Prevention of Premature Ejaculation, RENAL-UROLOGIC DRUGS, Treatment of Male Sexual Dysfunction, 5-HT Reuptake Inhibitors【研发状态】Phase III【合成情况】〖来源〗J Label Compd Radiopharm〖合成路线〗〖标题〗A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer〖合成方法〗In the original synthesis of the title compound, Knoevenagel condensation of benzaldehyde (I) with malonic acid (II) in the presence of ammonium acetate produced the beta-aminoacid (III). Reductive alkylation of the amino group of (III) with formaldehyde produced the dimethyl amine (IV). Then, Fischer esterification of (IV) with ethanolic HCl furnished the intermediate amino ester (V). Amino ester (V) was alternatively obtained by Michael addition of dimethylamine to ethyl cinnamate (VI).Reduction of the ester function of (V) provided amino alcohol (VII). The sodium alkoxide of (VII) was then coupled with 1-fluoronaphthalene (VIII) to produce the racemic amino ether, which was finally resolved into enantiomers by means of tartaric acid.〖作者〗Wheeler, W.J.; O'Bannon, D.D.〖参考〗Wheeler, W.J.; O'Bannon, D.D.; A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer. J Label Compd Radiopharm 1992, 31, 4, 305〖出处〗J Label Compd Radiopharm1992,31,(4):305〖来源〗J Label Compd Radiopharm〖合成路线〗〖标题〗A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer〖合成方法〗An alternative synthesis starting from the chiral precursor N-Boc-(R)-phenylglycine (IX) was reported. Borane reduction of (IX) provided the N-Boc aminoalcohol (X), which was activated as the mesylate (XI) by reaction with methanesulfonyl chloride in pyridine, yielding (XI). Displacement of the mesylate group of (XI) with NaCN furnished theBoc-aminonitrile (XII). Hydrolysis of the nitrile group of (XII) with concomitant N-Boc group cleavage under acidic conditions gave aminoacid (XIII). This was reduced toamino alcohol (XIV) using borane in THF. Eschweiler-Clarke methylation of aminoalcohol (XIV) yielded the dimethyl amine (XV). This was finally condensed with 1-fluoronaphthalene (VIII) to produce the title naphthyl ether. The [14C]-labeled compound was similarly prepared employing 14C-sodium cyanide.〖作者〗Wheeler, W.J.; O'Bannon, D.D.〖参考〗Wheeler, W.J.; O'Bannon, D.D.; A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer. J Label Compd Radiopharm 1992, 31, 4, 305〖出处〗J Label Compd Radiopharm1992,31,(4):305〖来源〗Nucl Med Biol〖合成路线〗〖标题〗Synthesis of [11C]dapoxetine.HCl, a serotonin re-uptake inhibitor: biodistribution in rat and preliminary PET imaging in the monkey〖合成方法〗The synthesis of the [11C]-labeled compound was also reported. Selective tosylation of the primary hydroxyl of (R)-1-phenyl-1,3-propanediol (XVI) provided (XVII). From this, naphthyl ether (XIX) was prepared by Williamson's synthesis with the sodium alkoxide of 1-naphthol (XVIII). The remaining hydroxyl group of (XIX) was then converted to mesylate (XX) upon treatment with methanesulfonyl chloride and DMAP. Subsequent displacement with methylamine in a sealed vessel afforded the secondary amine (XXI). This was finally alkylated with 11CH3I to yield the target 11C-labeled compound.〖作者〗Livni, E.; et al.〖参考〗Livni, E.; et al.; Synthesis of [11C]dapoxetine.HCl, a serotonin re-uptake inhibitor: biodistribution in rat and preliminary PET imaging in the monkey. Nucl Med Biol 1994, 21, 4, 669〖出处〗Nucl Med Biol1994,21,(4):669〖来源〗AU 8814335; EP 0288188; JP 1988258837; US 5135947〖合成路线〗〖标题〗1-Phenyl-3-naphthalenyloxypropanamines〖合成方法〗In the original synthesis of the title compound, Knoevenagel condensation of benzaldehyde (I) with malonic acid (II) in the presence of ammonium acetate produced the beta-aminoacid (III). Reductive alkylation of the amino group of (III) with formaldehyde produced the dimethyl amine (IV). Then, Fischer esterification of (IV) with ethanolic HCl furnished the intermediate amino ester (V). Amino ester (V) was alternatively obtained by Michael addition of dimethylamine to ethyl cinnamate (VI).Reduction of the ester function of (V) provided amino alcohol (VII). The sodium alkoxide of (VII) was then coupled with 1-fluoronaphthalene (VIII) to produce the racemic amino ether, which was finally resolved into enantiomers by means of tartaric acid.〖作者〗Robertson, D.W.; Thompson, D.C.; Wong, D.T. (Eli Lilly and Company)〖参考〗Robertson, D.W.; Thompson, D.C.; Wong, D.T. (Eli Lilly and Company);1-Phenyl-3-naphthalenyloxypropanamines. AU 8814335; EP 0288188; JP 1988258837; US 5135947〖出处〗AU 8814335; EP 0288188; JP 1988258837; US 5135947,,():。
达泊西汀合成总结

达泊西汀合成总结引言达泊西汀(Dapoxetine)是一种专门用于治疗早泄的选择性5-羟色胺再摄取抑制剂。
由于早泄是男性性功能障碍中最常见的问题之一,因此达泊西汀合成就显得非常重要。
本文将概述达泊西汀的合成过程,并介绍其中的关键步骤和反应条件。
合成步骤达泊西汀的合成过程通常基于对5-羟基色胺的一系列化学反应进行改进和改造。
下面是达泊西汀的一种合成方法,其中包括了主要的合成步骤和反应条件。
步骤一:取代氢化首先,将4-氯-3-硝基苯胺经过醚处理和盐酸处理,得到4-氯-3-硝基苯胺的盐酸盐。
然后,使用芳香胺类化合物对其进行取代反应,生成目标产物7-硝基-4-(3-取代基苄基)吡啶类化合物。
反应条件: - 取代反应中使用合适的芳香胺类化合物 - 利用合适的酸性催化剂步骤二:氢化还原目标产物经过氢化反应,将硝基还原为胺基。
该步骤通常在加氢反应器中进行,并且需要适当的压力和温度条件。
结果是得到7-胺基-4-(3-取代基苄基)吡啶化合物。
反应条件: - 反应温度:40-60°C - 反应压力:正常压力下加氢 - 加氢催化剂:常用的加氢催化剂如氢气和钯碳步骤三:酰化反应在这一步骤中,通过酰化反应将胺基化合物与2-(二甲基氨基)乙酸进行反应,生成目标产物达泊西汀的酰化衍生物。
反应条件: - 反应温度:室温 - 催化剂:具有酰化活性的催化剂,如二甲基亚锡(DMAP)步骤四:碱解反应在最后一步中,通过碱解反应使酰化衍生物脱去酰基,形成达泊西汀的自由胺。
反应条件: - 碱催化剂:常用的碱催化剂如碳酸钠或氢氧化钠 - 反应温度:室温结论通过这种合成路线,我们可以有效地合成达泊西汀。
根据文献报道,该方法可产率高,反应条件温和,适用于大规模合成。
然而,为了确保产品质量和纯度,仍需对合成过程进行完善和优化。
未来的研究可以尝试改进合成路径、催化剂和反应条件,以提高达泊西汀的合成效率和产率。
总之,达泊西汀作为一种用于治疗早泄的药物,在医疗领域发挥着重要的作用。
达泊西汀的合成进展

达泊西汀的合成进展
张飞皇;陈弢;徐云根
【期刊名称】《中国新药杂志》
【年(卷),期】2007(16)2
【摘要】达泊西汀是一类新型的、作用快速的选择性5-羟色胺再摄取抑制剂,早期用于治疗抑郁症和相关情感障碍,近年来则主要用于男性性功能障碍中早泄的治疗研究.因其化学稳定性好、起效迅速、不良反应相对较小等优点,已经越来越受到人们的关注.现对其4种合成方法进行了归纳和介绍,为该药的进一步研究提供参考.【总页数】3页(P111-113)
【作者】张飞皇;陈弢;徐云根
【作者单位】中国药科大学新药研究中心,南京,210009;常州市新北区生产力促进中心,常州,213022;中国药科大学新药研究中心,南京,210009
【正文语种】中文
【中图分类】R914.5;R971.43
【相关文献】
1.盐酸达泊西汀的合成 [J], 薛大泉;张威;邓泽军;马红梅;郑实;虞心红
2.达泊西汀关键中间体的合成 [J], 郭建敏;杨锐生;杜曦;王译伟
3.达泊西汀合成研究进展 [J], 王亚娜;陶莹莹;张恩
4.盐酸达泊西汀的合成与表征 [J], 王福生; 苏艳华
5.盐酸达泊西汀的合成及工艺优化 [J], 付丙月; 张宁; 张宗磊; 段崇刚
因版权原因,仅展示原文概要,查看原文内容请购买。
达泊西汀合成研究进展

( S c h o o l o f P h a r ma c e u t i c a l S c i e n c e Z h e n g z h o u Un i v e r s i t y, Zh e n g z h o u 4 5 0 0 0 1 , Ch i n a )
叭
达 泊 西汀药 物 的合成 方法 有 很多种 【 】 J ,归结 起来 可分 为两 大 类 :第 大类 是非 手性 的合 成方 法 ,即通 过得 到 外消旋 的 达泊 西 汀 或 者其 中间 体 ,然后 再进 行拆 分进 而得 到单 一 构 型 的达泊 西 汀 ;第二 大类 是不 对称 合成 方法 , 即通常 是 由金 属催 化或 生物 酶 合 成 方法 ,不 需要拆 分 直接 合成 构 型的达 泊西 汀 。这里 就按 照 这 两 个分 类分 别介 绍 。本 文就达 泊 西汀经 典 的合 成方 法进 行分 析 和讨论。
广
8 8
东
化
工
2 0 1 5年 第 6期
x v ww. g d c h e m. t o m
第4 2卷总第 2 9 6期
达 泊西 汀 合 成研 究 进 展
王 亚娜 ,陶 莹 莹 ,张 恩
( 郑 州大 学 药学 院 ,河南 郑 州 4 5 0 0 0 1 )
Re s e a r c h Pr o gr e s s o f Da po x i t i n e Sy nt h e t i c Me t ho d
A b s t r a c t : D a p o x e t i n e i s t h e w o r l d ’ S i f r s t a p p r o v e d o r a l me d i c a t i o n f o r t h e t r e a t me n t o f p r e ma t u r e e j a c u l a t i o n . B e c a u s e o f h a v i n g a c h i r a l c e n t e r , a n d t h e r e f o r e
盐酸达泊西汀的合成及工艺优化

盐酸达泊西汀的合成及工艺优化作者:付丙月张宁张宗磊段崇刚来源:《中国药房》2020年第07期摘要目的:优化盐酸达泊西汀的合成工艺。
方法:采用手性合成的方法,以3-氯苯丙酮为原料,采用(1S,2R)-(-)-1-氨基-2-茚醇为催化剂、硼烷-N,N-二乙基苯胺为还原剂进行不对称还原,然后依次经过与α-萘酚醚化、磺酸酯化、二甲胺取代、HCl成盐反应得到最终产物。
通过核磁共振和质谱技术对合成产物进行表征。
对中间体Ⅰ、中间体Ⅱ、中间体Ⅲ和最终产物的合成反应进行工艺优化。
结果:表征结果显示最终产物为盐酸达泊西汀,纯度为99.8%,收率为58.9%。
与传统的拆分工艺比较,本工艺采用手性合成的方法不需要拆分,收率明显高于文献报道的拆分工艺收率(31.9%)。
优化后的工艺减少了杂质的产生,提高了产品质量。
结论:本合成工艺反应条件较温和、合成路线较短、收率较高。
关键词盐酸达泊西汀;手性合成;不对称还原;工艺优化ABSTRACT; ;OBJECTIVE: To optimize the synthesis process of dapoxetine hydrochloride. METHODS: By chiral synthesis, asymmetric reduction was carried out by using 3-chlorophenylacetone as raw material,(1S, 2R) -(-)-1-amino-2-indanol as catalyst, and borane-N, N-diethylaniline (DEANB) as reducing agent. Then,it was reacted with α-naphthol etherification, sulfonation, dimethylamine substitution, and HCl salt formation reaction to obtain the final products. The products were characterized by NMR and MS. The synthesis reaction of intermediate Ⅰ, intermediate Ⅱ, intermediate Ⅲ and the final product were optimized. RESULTS: The final product was dapoxetine hydrochloride with purity of 99.8% and yield of 58.9%. Compared with traditional splitting technology, the chiral synthesis technology of this study did not need splitting, and the yield of the technology was significantly higher than that of splitting technology reported in literature (31.9%). The optimized technology reduced the generation of impurities and improved the product quality. CONCLUSIONS: The improved technology has milder reaction conditions, shorter synthesis route and higher yield.KEYWORDS; ;Dapoxetine hydrochloride; Chiral synthesis; Asymmetric reduction; Technology optimization鹽酸达泊西汀(Dapoxetine hydrochloride)化学名为(S)-(+)-(N,N-二甲胺基)-3-(萘基-1-氧基)-1-苯基丙烷盐酸盐,是一种选择性5-羟色胺再摄取抑制剂(SSRIs),2009年在欧洲上市,之后陆续在多个国家和地区上市,2013年进入中国市场(商品名:必利劲),用于成年男子早泄的按需治疗,是世界上第一个被批准用于治疗早泄的口服药[1]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
[关键词 ] 达泊西汀 ;抗抑郁 ;早泄 ;药物合成 [中图分类号 ] R914. 5; R971. 43 [文献标识码 ] A [文章编号 ] 1003 - 3734 (2007) 02 - 0111 - 03
Syn thesis of dapoxetine
ZHANG Fei2huang1 , CHEN Tao2 , XU Yun2gen1 ( 1 Cen ter of D rug D iscovery, Ch ina Pha rm aceu tica l U n iversity, N an jing 210009, Ch ina; 2 X inbei D istrict of Changzhou C ity P roductivity P rom otion Cen ter, Changzhou 213022, Ch ina)
中国新药杂志 2007年第 16卷第 2期
图 1 达泊西汀的结构式
达泊西汀的合成方法ห้องสมุดไป่ตู้达泊西汀的合成方法按起始原料的不同 ,可分
为以下 4种 。 111 以苯甲醛和丙二酸为原料 苯甲醛和丙二酸 经过克脑温格尔缩合反应生成 β2氨基苯丙酸 ( 2) , 继而其氨基用甲醛还原烃化得到 32(二甲氨基 ) 232 苯基丙酸 (3)后将其制成乙酯 4 ( Scheme 1) 。4也可 直接以肉桂酸乙酯为原料 ,与二甲胺通过迈克尔加成 获得 。然后 , 4用二 (22甲氧基乙氧基 )氢化铝或者四 氢锂铝还原得到氨基醇 (5) , 5在氢化钠作用下与 12 氟萘反应生成达泊西汀消旋体 ( 6) ,最后用 L 2( + ) 2 酒石酸拆分就可得到本品 (1) [4, 5] ,合成路线见图 2。
图 3 方法 2的合成路线
113 直接以手性化合物 N 2Boc2( R ) 2苯基甘氨酸为 原料 手性化合物 N 2Boc2( R ) 2苯基甘氨酸用硼烷 还原得氨基醇 ( 9 ) , 9 和甲磺酰氯反应得甲磺酸酯 (10) ,再与氰化钠反应得氰基物 ( 11) , 11在酸性条 件下水解同时脱去 N 上保护基 Boc得到手性 β2氨 基酸 (12) , 12用硼烷还原得羟基物 ( 13) , 13 经 Es2
[ Key words] dapoxetine; antidep ressant; p rem ature ejaculation; synthesis
达泊西汀 ( dapoxetine, LY2210448, 1 )是一个选 择性的 52羟色胺再摄取抑制剂 ,结构上类似于氟西 汀并同样具有抗抑郁作用 ,临床研究用其盐酸
— 112 —
图 4 方法 3的合成路线
114 以 ( R ) 212苯基 21, 32丙二醇为原料 用对甲基 苯磺酰氯对 ( R ) 212苯基 21, 32丙二醇选择性磺酰化 羟基得 15, 15 和 12萘 酚 缩 合 得 到 ( R ) 232( 12萘 氧 基 ) 212苯基丙醇 16 ( Scheme 5) 。化合物 16 也可以 用 (R ) 232氯 212苯 基 丙 醇 和 12萘 酚 缩 合 反 应 得 到 ( Scheme 6) 。然后 16 和甲磺酰氯反应生成甲磺酰
图 2 方法 1的合成路线
112 以 12萘酚和 32苯基丙基溴为原料 12萘酚和 chweiler2C larke甲基化后生成二甲胺物 ( 14 ) , 14 与 32苯基丙基溴经过威廉姆生醚合成得到 12苯基 232 12氟萘缩合即得到本品 [5 ] ,合成路线见图 4。 (12萘氧基 )丙烷 ( 7) ,继而用 NBS溴代得到苄位溴 代物 ( 8) , 8和二甲胺反应 ,生成 ( 6) ,最后拆分得到 本品 [ 5 ] ,合成路线见图 3。
with top iramate monotherapy in elderly patients with recent2onset ep ilep sy[ J ]. A cta N eu rol S cand, 2005, 112 ( 3) : 144 - 150. [ 14 ] KAL IN IN VV , ZHELEZNOVA EV , SOKOLOVA LV , et a l. To2 pamax monotherapy of partial ep ilep sy[ J ]. Zh N evropa tol Psikhi2 atr, 2004, 104 ( 7) : 35 - 38. [ 15 ] BERG AT, SH INNAR S, LEVY SR, et a l. How well can ep ilep sy syndromes be identified at diagnosis? A reassessment 2 years after initial diagnosis[ J ]. Epilepsia, 2000, 41 ( 10) : 1269 - 1275. [ 16 ] KWAN P, BROD IE MJ. Effectiveness of first antiep ilep tic drug [ J ]. Epilepsia, 2001, 42 ( 10) : 1255 - 1260. [ 17 ] CROSS JH. Top iramate monotherapy for childhood absence sei2 zures: an open label p ilot study[ J ]. S eizure, 2002, 11 ( 6) : 406 410. [ 18 ] PR IV ITERA MD , BROD IE MJ, MATTSON RH , et a l. Top ira2 mate, carbamazep ine and valp roate monotherapy: double2blind comparison in newly diagnosed ep ilep sy[ J ]. A cta N eu rol S cand,
中国新药杂志 2007年第 16卷第 2期 2003, 107 ( 3) : 165 - 175. [ 19 ] SILBERSTE IN SD , BEN 2M ENACHEM E, SHANK RP, et a l. Top iramate monotherapy in ep ilep sy and m igraine p revention [ J ]. C lin Ther, 2005, 27 ( 2) : 154 - 165. [ 20 ] VALENC IA I, FONS C, KOTHARE SV , et al. Efficacy and toler2 ability of top iramate in children younger than 2 years old [ J ]. J Ch ild N eu rol, 2005, 20 ( 8) : 667 - 669. [ 21 ] KARCESKI S, MORRELL MJ, CARPENTER D. Treatment of ep ilep sy in adults: expert op inion, 2005 [ J ]. Epilepsy B ehav, 2005, ( Supp l 1) : S1 - S64; quiz S65 - S67. [ 22 ] FRENCH JA , KANNER AM , BAUTISTA J, et a l. Efficacy and tolerability of the new antiep ilep tic drugs I: treatment of new on2 set ep ilep sy: report of the Therapeutics and Technology A ssess2 ment Subcomm ittee and Quality Standards Subcomm ittee of the American Academy of Neurology and the American Ep ilep sy Soci2 ety[ J ]. N eurology, 2004, 62 ( 8) : 1252 - 1260.
编辑 :杨青 /接受日期 : 2006 - 09 - 10
达泊西汀的合成进展
张飞皇 1 , 陈 弢 2 , 徐云根 1 (1 中国药科大学新药研究中心 ,南京 210009; 2 常州市新北区生产力促进中心 ,常州 213022)
[摘要 ] 达泊西汀是一类新型的 、作用快速的选择性 52羟色胺再摄取抑制剂 ,早期用于治疗抑郁症和相关 情感障碍 ,近年来则主要用于男性性功能障碍中早泄的治疗研究 。因其化学稳定性好 、起效迅速 、不良反应相对 较小等优点 ,已经越来越受到人们的关注 。现对其 4 种合成方法进行了归纳和介绍 ,为该药的进一步研究提供 参考 。
Chinese Journal of New D rugs 2007, Vol. 16 No. 2 [ 12 ] W ILLMORE LJ. Management of ep ilep sy in the elderly[ J ]. Epi2
lepsia, 1996, 37 ( Supp l 6) : S23 - S33. [ 13 ] GROSELJ J, GUERR IN I R , VAN OENE J, et a l. Experience