达泊西汀的合成进展
盐酸达泊西汀的合成

2.2
1-苯基-3-(1-萘氧基)-1-丙醇的制备(4) 在 250 mL 三口烧瓶中加入化合物 3(10.00 g,
副产物的产生。经醚化反应制备化合物 4 时,文 献 [3]采用溶剂二甲基亚砜,催化剂碳酸钾,反应 温度为 90 ℃,原料反应不完全,且副产物多,需 进行纯化才能投入下一步反应。本研究采用二甲 基甲酰胺作溶剂,氢氧化钠作催化剂,温度降低 到 50 ℃ , 改 进 后 原 料 反 应 完 全 且 纯 度 提 升 到>98%,不需纯化后处理,可直接投入下一步反 应,简化了操作步骤。经亲核取代反应制备化合 物 5 时, 文献[3]采用 4-二甲氨基吡啶(DMAP)作催 化剂,但 DMAP 在后处理中很难除去,残留在产 品中成为一种杂质,实验证明不需要添加 DMAP 也可反应完全。此步反应加入甲磺酰氯时放热剧 烈,温度过高容易产生副产物,本研究将反应温 度由 5~0 ℃降低到 10~5 ℃,能更好的控制温 度,减少副产物的生成。文献[3]在加入二甲胺后 反应时间为 40 h,笔者研究发现 20 h 即可反应完 毕,可大大缩短反应时间。改进方法后纯度达到 97%。化合物 5 经 D-(+)-DTTA 拆分时,按照文献 [3]采用二氯甲烷作溶剂、拆分温度为 25~35 ℃, 收率在 28%左右。在采用二氯甲烷作溶剂的基础 上,加入一定量的乙酸乙酯作反溶剂,并将拆分 温度降至 15 ℃, 能将收率提高到>43%。 达泊西汀 DTTA 盐经氢氧化钠溶液游离, 成盐酸盐得到最终 目的产物。在纯化达泊西汀盐酸盐的这一步骤中, 文献[3]采用 10~15 ℃析晶,重复操作发现在此温 度下重结晶收率仅在 60%左右,改用8 ℃析晶收 率能提高到 >90% ,纯度达到 99.98% ,最大单杂 <0.01%。改进后的工艺原料易得,操作简便,更 利于工业化生产。消旋体收率达到 91%,总收率 达到 35%,比已报道的文献值有了较大提高。 2 2.1 实验部分 3-氯苯丙醇(3)的制备 在 250 mL 三口烧瓶中加入化合物 2(10 g,
达泊西汀的合成进展_张飞皇

[关键词 ] 达泊西汀 ;抗抑郁 ;早泄 ;药物合成 [中图分类号 ] R914. 5; R971. 43 [文献标识码 ] A [文章编号 ] 1003 - 3734 (2007) 02 - 0111 - 03
Syn thesis of dapoxetine
ZHANG Fei2huang1 , CHEN Tao2 , XU Yun2gen1 ( 1 Cen ter of D rug D iscovery, Ch ina Pha rm aceu tica l U n iversity, N an jing 210009, Ch ina; 2 X inbei D istrict of Changzhou C ity P roductivity P rom otion Cen ter, Changzhou 213022, Ch ina)
中国新药杂志 2007年第 16卷第 2期
图 1 达泊西汀的结构式
达泊西汀的合成方法ห้องสมุดไป่ตู้达泊西汀的合成方法按起始原料的不同 ,可分
达泊西汀药物制备技术研究进展

作者简 介: 张丽萍( 1 9 9 O 一 ) , 女, 硕士研究生, 研究方 向: 生物催化与手性合成 。E - m a i l : l p z h a n g 8 0 9 0 @1 6 3 . c o m
通讯作者 : 王普 , 女, 教授 , 博 士 生导 师 。
一
1 8一
Z H E J I A N G C H E MI C A L I N D U S T R Y
多 ,仅 需 添 加 少 量 手 性 催 化 剂 ( 二磷 配体与铱 、
铑、 钌 的络化物等 ) 即可得 到 大量手性 化合物 , 且
R构 型 和 S构 型 均 易 获 得 l 5 l 。 虽 然 手 性 催 化 剂 专 一
比, 达泊 西汀具有 疗效好 、 半衰期 短 、 副作用 少等
1 化 学 不 对 称 合 成 法
化 学 催 化 不 对 称 合 成 是 制 备 手 性 化 合 物 的 重 要方 法 , 它 通过 特定 的手性催 化 剂 。 将 潜 手 性 底 物 转 化 为单 一 构 型 的 光 学 纯 化 合 物 。该 方 法 广
泛运 用于手性 化合物 的不对称 合成 。 反 应 类 型 众
V o 1 . 4 6 N o . 6 ( 2 0 1 5 )
反 应 步 骤 繁 琐 ,因 此 在 工业 生 产 上 能 有 效 应 用 的
DMF ( 2 : 1 ) 体 系 中 发 生 亲 核 取 代 反应 生 成 ( 6 ) , ( 6 ) 在钯金属催化 剂的作用下 氢化 , 脱 去 氨 基 甲酸 酯 保护 , 得 到化 合物 ( 7 ) , 最后 ( 7 ) 发生 E s c h w e i l e r — C l a r k e反 应 得 到 ( 1 ) , 总收率为 2 8 . 5 %。该 方 法 合 成路线 较长 , 收率偏 低 , 且 使 用 甲苯 和钯金 属催 化剂等有毒物 质 , 因 此 改 进 此 合 成 路 线 成 为 研 究 热 点 。 Ma h a l e等 嘲 以手性 恶唑硼 烷 为催化 剂 , 在 D E AN B体 系 中不 对 称 还 原 ( 2 ) 得到( R) 一 3 一 氯 苯
盐酸达泊西汀生产工艺

盐酸达泊西汀生产工艺盐酸达泊西汀是一种常用的化学药品,它在医药领域有着广泛的应用。
本文将介绍盐酸达泊西汀的生产工艺。
盐酸达泊西汀的生产工艺主要分为以下几个步骤。
首先,将原料达泊西汀与盐酸溶液进行反应。
这一步骤的目的是将达泊西汀与盐酸结合,形成盐酸达泊西汀。
反应过程中需要注意控制温度和反应时间,以确保反应的完全性和产物的纯度。
接下来,对反应混合物进行过滤和干燥处理。
通过过滤,可以去除反应中产生的杂质和固体物质。
干燥处理可以将产物中的水分去除,提高产物的纯度和稳定性。
然后,对干燥后的产物进行结晶处理。
结晶过程中,可以通过控制温度和溶剂的浓度来促使产物结晶。
结晶后的产物通常具有较高的纯度和良好的结晶形态。
对结晶后的产物进行干燥和粉碎处理。
干燥可以进一步去除残留的水分,提高产物的稳定性和保存期限。
粉碎处理可以将产物研磨成所需的粒度,以满足不同的应用需求。
盐酸达泊西汀的生产工艺中,需要注意的一些关键技术点。
首先,反应温度和反应时间的控制十分重要,这可以确保反应的完全性和产物的纯度。
其次,过滤和干燥处理需要采用适当的方法和设备,以提高产物的纯度和稳定性。
此外,结晶过程中的温度和溶剂浓度的选择也对产物的纯度和结晶形态有着重要影响。
盐酸达泊西汀的生产工艺需要严格遵守相关的安全操作规程。
在操作过程中,应注意防止产生有害气体和溶液的泄漏。
同时,应配备必要的防护设施,如安全眼镜、手套和防护服等,以确保操作人员的安全。
盐酸达泊西汀的生产工艺是一个复杂而严谨的过程。
通过控制反应条件、过滤干燥和结晶处理等关键步骤,可以获得高纯度和良好结晶形态的盐酸达泊西汀产物。
在实际生产中,应严格遵守相关的操作规程和安全要求,确保产品质量和操作人员的安全。
达泊西汀(dapoxetine)路线
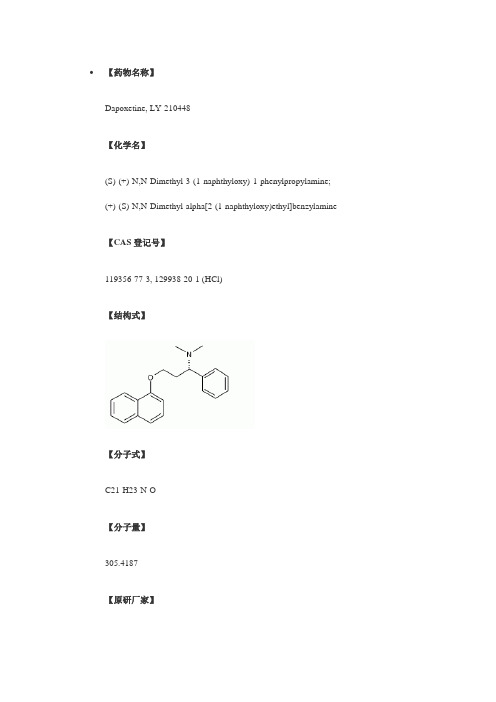
【药物名称】Dapoxetine, LY-210448【化学名】(S)-(+)-N,N-Dimethyl-3-(1-naphthyloxy)-1-phenylpropylamine;(+)-(S)-N,N-Dimethyl-alpha[2-(1-naphthyloxy)ethyl]benzylamine 【CAS登记号】119356-77-3, 129938-20-1 (HCl)【结构式】【分子式】C21-H23-N-O【分子量】305.4187【原研厂家】Lilly (Originator), Dynogen Pharmaceuticals (Not Determined), Alza (Licensee), PPD GenuPro (Licensee)【作用类别】Prevention of Premature Ejaculation, RENAL-UROLOGIC DRUGS, Treatment of Male Sexual Dysfunction, 5-HT Reuptake Inhibitors【研发状态】Phase III【合成情况】〖来源〗J Label Compd Radiopharm〖合成路线〗〖标题〗A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer〖合成方法〗In the original synthesis of the title compound, Knoevenagel condensation of benzaldehyde (I) with malonic acid (II) in the presence of ammonium acetate produced the beta-aminoacid (III). Reductive alkylation of the amino group of (III) with formaldehyde produced the dimethyl amine (IV). Then, Fischer esterification of (IV) with ethanolic HCl furnished the intermediate amino ester (V). Amino ester (V) was alternatively obtained by Michael addition of dimethylamine to ethyl cinnamate (VI).Reduction of the ester function of (V) provided amino alcohol (VII). The sodium alkoxide of (VII) was then coupled with 1-fluoronaphthalene (VIII) to produce the racemic amino ether, which was finally resolved into enantiomers by means of tartaric acid.〖作者〗Wheeler, W.J.; O'Bannon, D.D.〖参考〗Wheeler, W.J.; O'Bannon, D.D.; A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer. J Label Compd Radiopharm 1992, 31, 4, 305〖出处〗J Label Compd Radiopharm1992,31,(4):305〖来源〗J Label Compd Radiopharm〖合成路线〗〖标题〗A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer〖合成方法〗An alternative synthesis starting from the chiral precursor N-Boc-(R)-phenylglycine (IX) was reported. Borane reduction of (IX) provided the N-Boc aminoalcohol (X), which was activated as the mesylate (XI) by reaction with methanesulfonyl chloride in pyridine, yielding (XI). Displacement of the mesylate group of (XI) with NaCN furnished theBoc-aminonitrile (XII). Hydrolysis of the nitrile group of (XII) with concomitant N-Boc group cleavage under acidic conditions gave aminoacid (XIII). This was reduced toamino alcohol (XIV) using borane in THF. Eschweiler-Clarke methylation of aminoalcohol (XIV) yielded the dimethyl amine (XV). This was finally condensed with 1-fluoronaphthalene (VIII) to produce the title naphthyl ether. The [14C]-labeled compound was similarly prepared employing 14C-sodium cyanide.〖作者〗Wheeler, W.J.; O'Bannon, D.D.〖参考〗Wheeler, W.J.; O'Bannon, D.D.; A chiral synthesis of dapoxetine hydrochloride, a serotonin reuptake inhibitor, and its 14C isotopomer. J Label Compd Radiopharm 1992, 31, 4, 305〖出处〗J Label Compd Radiopharm1992,31,(4):305〖来源〗Nucl Med Biol〖合成路线〗〖标题〗Synthesis of [11C]dapoxetine.HCl, a serotonin re-uptake inhibitor: biodistribution in rat and preliminary PET imaging in the monkey〖合成方法〗The synthesis of the [11C]-labeled compound was also reported. Selective tosylation of the primary hydroxyl of (R)-1-phenyl-1,3-propanediol (XVI) provided (XVII). From this, naphthyl ether (XIX) was prepared by Williamson's synthesis with the sodium alkoxide of 1-naphthol (XVIII). The remaining hydroxyl group of (XIX) was then converted to mesylate (XX) upon treatment with methanesulfonyl chloride and DMAP. Subsequent displacement with methylamine in a sealed vessel afforded the secondary amine (XXI). This was finally alkylated with 11CH3I to yield the target 11C-labeled compound.〖作者〗Livni, E.; et al.〖参考〗Livni, E.; et al.; Synthesis of [11C]dapoxetine.HCl, a serotonin re-uptake inhibitor: biodistribution in rat and preliminary PET imaging in the monkey. Nucl Med Biol 1994, 21, 4, 669〖出处〗Nucl Med Biol1994,21,(4):669〖来源〗AU 8814335; EP 0288188; JP 1988258837; US 5135947〖合成路线〗〖标题〗1-Phenyl-3-naphthalenyloxypropanamines〖合成方法〗In the original synthesis of the title compound, Knoevenagel condensation of benzaldehyde (I) with malonic acid (II) in the presence of ammonium acetate produced the beta-aminoacid (III). Reductive alkylation of the amino group of (III) with formaldehyde produced the dimethyl amine (IV). Then, Fischer esterification of (IV) with ethanolic HCl furnished the intermediate amino ester (V). Amino ester (V) was alternatively obtained by Michael addition of dimethylamine to ethyl cinnamate (VI).Reduction of the ester function of (V) provided amino alcohol (VII). The sodium alkoxide of (VII) was then coupled with 1-fluoronaphthalene (VIII) to produce the racemic amino ether, which was finally resolved into enantiomers by means of tartaric acid.〖作者〗Robertson, D.W.; Thompson, D.C.; Wong, D.T. (Eli Lilly and Company)〖参考〗Robertson, D.W.; Thompson, D.C.; Wong, D.T. (Eli Lilly and Company);1-Phenyl-3-naphthalenyloxypropanamines. AU 8814335; EP 0288188; JP 1988258837; US 5135947〖出处〗AU 8814335; EP 0288188; JP 1988258837; US 5135947,,():。
盐酸达泊西汀的合成工艺研究_尹玲丽
收稿日期:2010-09-29作者简介:尹玲丽(1985-),女(汉族),浙江台州人,硕士研究生,E m a i:l y i nli ng li @163.co m;*通讯作者:陈国华(1963-),男(汉族),福建莆田人,副研究员,硕士生导师,主要从事药物化学研究,T e:l (025)83241246,E m a i:l cgh63@163.co m 。
文章编号:1005-0108(2011)01-0037-03盐酸达泊西汀的合成工艺研究尹玲丽,陈国华*(中国药科大学药物化学教研室,江苏南京210009)摘 要:目的研究选择性5 羟色胺重摄取抑制剂盐酸达泊西汀的合成工艺。
方法以3 苯基丙醇和1 氟萘为起始原料,经醚化、溴代、二甲胺基取代、拆分、成盐反应制得目标化合物。
结果与结论目标化合物的结构经1H NM R 、M S 、I R 谱以及比旋光度确证。
该路线原料易得,操作简便,条件温和,有利于工业化生产,消旋体收率达61 2%。
关键词:选择性5 羟色胺重摄取抑制剂;盐酸达泊西汀;化学合成;工艺改进中图分类号:R 914 文献标志码:A盐酸达泊西汀(dapoxetine hydrochlori d e ,1)化学名为(S ) (+) (N,N 二甲胺基) 3 (萘基 1 氧基) 1 苯基丙烷盐酸盐,是一种选择性5 羟色胺重摄取抑制剂(SSRI),由美国礼来制药公司(E li L ill y )研制,2009年在欧洲上市,商品名为Prili g y ,用于治疗男性早泄(PE)。
该药半衰期短、不良反应小、效果显著,是世界上第一种被批准治疗PE 的经口给药的处方药[1]。
1 合成路线文献报道的合成盐酸达泊西汀(1)的方法较多[2-7]。
本研究参考文献[2]的方法,以3 苯丙醇(2)、1 氟萘(3)为起始原料,在氢化钠作用下成醚得到化合物4,4经NBS 苄位溴代得到5,化合物5与二甲胺进行亲核取代反应得到化合物6,6经L (+) 酒石酸拆分,成盐酸盐得目标化合物1。
盐酸达泊西汀的合成与表征

1 实验部分
1.1 仪器及试剂
Vario EL Ⅲ元素分析仪,Nexus 型傅里叶变换 红外 (FT-IR) 分析仪,Waters SQD2 液相色谱质谱联 用仪,Avance Ⅲ 400MHz 核磁共振谱仪,WZZ-2B 型自动旋光仪。
苯丙醇、三氟乙酸酐、三氟化硼乙醚、乙腈、乙酸 乙酯、NBS、偶氮二异丁腈、四氢呋喃、二甲胺四氢呋 喃溶液、三乙胺、二氯甲烷、氯仿、无水硫酸钠、氢氧 化钠、DTTA、异丙醇、氯化氢。
盐酸达泊西汀化学名为 (S)-(+)-N,N- 二甲基 -3-
(1- 萘氧基 )-1- 苯基丙胺盐酸盐,是一种选择性 5-
羟胺再摄取抑制剂,具有半衰期短、不良反应较小等
优点,由美国礼来制药公司研制并于 2009 年在欧洲
上市,用于治疗男性早泄。 国内外报道的盐酸达泊西汀的合成文献较多 , [1-6]
OOH 1) (F3CCO)2O
2)
OH
O NBS , AIBN
Br O
2
3
4
5
HN(CH3)2
N O 1) DTTA 2) NaOH
N O HCl
N ·HCl O
6
7
1
图 1 盐酸达泊西汀的合成路线 Fig.1 Synthetic route of dapoxetine hydrochloride
大后危险性较高。本文采用拆分法,以苯丙醇 (2) 为
起始原料,先后依次与三氟乙酸酐、1- 萘酚反应生成
化合物 4,化合物 4 再经 NBS 溴代得到化合物 5,化
合物 5 与二甲胺进行亲核取代反应得到化合物 6,6
经 D-(+)- 二对甲基苯甲酰酒石酸拆分,成盐酸盐后
得到目标化合物 1。该合成路线见图 1。
达泊西汀合成总结

达泊西汀合成总结引言达泊西汀(Dapoxetine)是一种专门用于治疗早泄的选择性5-羟色胺再摄取抑制剂。
由于早泄是男性性功能障碍中最常见的问题之一,因此达泊西汀合成就显得非常重要。
本文将概述达泊西汀的合成过程,并介绍其中的关键步骤和反应条件。
合成步骤达泊西汀的合成过程通常基于对5-羟基色胺的一系列化学反应进行改进和改造。
下面是达泊西汀的一种合成方法,其中包括了主要的合成步骤和反应条件。
步骤一:取代氢化首先,将4-氯-3-硝基苯胺经过醚处理和盐酸处理,得到4-氯-3-硝基苯胺的盐酸盐。
然后,使用芳香胺类化合物对其进行取代反应,生成目标产物7-硝基-4-(3-取代基苄基)吡啶类化合物。
反应条件: - 取代反应中使用合适的芳香胺类化合物 - 利用合适的酸性催化剂步骤二:氢化还原目标产物经过氢化反应,将硝基还原为胺基。
该步骤通常在加氢反应器中进行,并且需要适当的压力和温度条件。
结果是得到7-胺基-4-(3-取代基苄基)吡啶化合物。
反应条件: - 反应温度:40-60°C - 反应压力:正常压力下加氢 - 加氢催化剂:常用的加氢催化剂如氢气和钯碳步骤三:酰化反应在这一步骤中,通过酰化反应将胺基化合物与2-(二甲基氨基)乙酸进行反应,生成目标产物达泊西汀的酰化衍生物。
反应条件: - 反应温度:室温 - 催化剂:具有酰化活性的催化剂,如二甲基亚锡(DMAP)步骤四:碱解反应在最后一步中,通过碱解反应使酰化衍生物脱去酰基,形成达泊西汀的自由胺。
反应条件: - 碱催化剂:常用的碱催化剂如碳酸钠或氢氧化钠 - 反应温度:室温结论通过这种合成路线,我们可以有效地合成达泊西汀。
根据文献报道,该方法可产率高,反应条件温和,适用于大规模合成。
然而,为了确保产品质量和纯度,仍需对合成过程进行完善和优化。
未来的研究可以尝试改进合成路径、催化剂和反应条件,以提高达泊西汀的合成效率和产率。
总之,达泊西汀作为一种用于治疗早泄的药物,在医疗领域发挥着重要的作用。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
1.2 以1-萘酚和3一苯基丙基溴为原料 1一萘酚和
chweiler—Clarke q|基化后生成二甲版物(14),14与
3一苯基丙基溴经过威廉姆生醚合成得到1一苯基.3一 1一氟萘缩合即得到本品…,合成路线见图4。
(1.萘氧基)丙烷(7),继而用NBS溴代得到苄位溴
代物(8),8和二甲胺反应,生成(6),最后拆分得到
[Abstract]Dapoxetine,a novel fast—acting selective serotonin reuptake inhibitor(SSRI),is used
to treat depression and affective disorder at preliminary stage,but nOW is mainly for the treatment of pros— permia which is one of the most commonly male sexual dysfunction.This drug is getting more popular due to better chemical stability,fast—acting property and little side effects.This paper reviews the synthetic methods of dapoxetine.
1.1 以苯甲醛和丙二酸为原料 苯甲醛和丙二酸
为治疗早泄药物,其半衰期短,不良反应小,效果
经过克脑温格尔缩合反应生成13一氨基苯丙酸(2),
显著"1。分析家预言达泊西汀的峰销售额可达到
继而其氨基用甲醛还原烃化得到3-(二甲氨基)一3.
5亿美元。虽然ALZA公司曾于2005年10月27
苯基丙酸(3)后将其制成乙酯4(Scheme 1)。4也可
pamax monotherapy bf partial epilepsy[J].Zh Nevropatol Psikhi-
atr,2004,104(7):35—38.
[15]
BERG AT,SHINNAR S,LEVY SR,et a1.How well can epilepsy
syndromes be identified at diagnosis?A reassessment 2 years after
H3C-,N,CH3 i
西飞 1
中心进行Ⅲ期临床试验‘2。。
图1达泊西汀的结构式
2005年5月,强生公司在100届全美泌尿协会
1达泊西汀的合成方法
科学会议(AUA.2005)期间公布了达泊西汀有关治
达泊西汀的合成方法按起始原料的不同,可分
疗早泄的Ⅲ期临床试验数据,同时也公布了其药物
为以下4种。
相互作用和药效学试验结果。各项指标均显示作
[21] KARCESKI S,MORRELL MJ,CARPENTER D.Treatment of
epilepsy in adults:expert Opinion,2005[J].Epilepsy Behav,
[22]
2005,(Suppl 1):S1一¥64;quiz¥65一¥67. FRENCH JA,KANNER AM,BAUTISTA J,et a1.Efficacy and
学研究工作。联系电话:(025)85339748,E—mail:xy964@
126.com。
[ 参考文献 ]
[1] RElD LR,RoBERT50N DW,THoMP50N Dc·E‰。ts。‘8‘。”一
8。嘣。nin(5HT)“9‘8。8。8m8山7
。chemistry—on.inhibition。‘the
[摘要] 达泊西汀是一类新型的、作用快速的选择性5一羟色胺再摄取抑制剂,早期用于治疗抑郁症和相关
情感障碍,近年来则主要用于男性性功能障碍中早泄的治疗研究。因其化学稳定性好、起效迅速、不良反应相对
较小等优点,已经越来越受到人们的关注。现对其4种合成方法进行了归纳和介绍,为该药的进一步研究提供
参考。 [关键词] 达泊西汀;抗抑郁;早泄;药物合成
[中圈分类号]R914.5;R971.43
[文献标识码]A [文章编号]1003—3734(2007)02—0111—03
Synthesis of dapoxetine
ZHANG Fei.huan91,CHEN Ta02,XU Yun—genl
(1 Center of Drug Discovery,China Pharmaceutical University,Nanjing 2 1 0009,China; 2 Xinbei District of Changzhou City Productivi钞Promotion Center,Changzhou 2 1 3022,China)
盐…。达泊西汀最初由美国礼来制药公司(Eli Lilly) 研制并作为抗抑郁药进行了I期临床试验,但其抗抑 郁作用并没有得到人们的一致认可;后该药归PPD
一111—
竺!!!!!!!!!翌型!!壁!兰望型g!!!!!!!!!:!!垡!:!
中国新药杂志2007年第16卷第2期
制药公司所有,并在其子公司PPD GenuPro指导下 作为治疗早泄药进行了II期临床试验;最后该药完 全归强生公司(Johnson&Johnson)的子公司阿尔扎 (ALZA)所有,并作为治疗早泄药在美国60个医疗
with topiramate monotherapy in elderly patients with recent—onset
epilepsy[J].Acta Neurol Scand,2005,112(3):144—150.
[14]
KALININ VV,ZHELEZNOVA EV,SOKOLOVA LV,et a1.To-
图3方法2的合成路线
1.3直接以手性化合物N-Boc-(冠)-苯基甘氨酸为 原料手性化合物N—Boc一(R)-苯基甘氨酸用硼烷 还原得氨基醇(9),9和甲磺酰氯反应得甲磺酸酯 (10),再与氰化钠反应得氰基物(11),11在酸性条 件下水解同时脱去N上保护基Boc得到手性B.氨 基酸(12),12用硼烷还原得羟基物(13),13经Es一
OMs
(o的 洲b
飞+∞ 16 oH
C
17
?一
NaH —————————●-16
OrNaOH
图5方法4的合成jl胥线
2 对合成方法的评价 方法1的起始原料苯甲醛和丙二酸价廉易得,
的合成研究,通过工艺改进,使之更适合于工业化生
产,所得产品的收率较高,纯度较好。
[作者简介] [通讯作者]
张飞皇(1981、),男,硕士研究生。 徐云根(1964一),男,教授,主要从事药物化
initial diagnosis[J].Eeilepsia,2000,41(10):1269—1275.
[16] KWAN P,BRODIE MJ.Effectiveness of first antiepileptic drug
[J].印ilepsia,2001,42(10):1255—1260.
一1 1 2一
万方数据
竖!i!!!!』!!!型!!型!!些翌g!!业:!!!!!:!!型!:!
化产物17,17和二甲胺反应得到目标产物¨’7|,合
成一路线。见图√5。固学b。√国鬲NaOH
主垦堑堑苤查!!!!生笙!!鲞筮!塑
3 结语 达泊西汀最早作为一个抗抑郁药上市,但现在
人们更期待它治疗早泄的新适应证能够获得FDA 的批准。本实验室选择上述方法2进行了达?白西汀
氟萘反应生成达泊西汀消旋体(6),最后用上-(+)一
图1。
酒石酸拆分就可得到本品(1)f4,51,合成路线见图2。
CHO+H碟一一。飞罟H。
ElOH
HCIEBiblioteka O【MeO(CH2)20]2AIH
Or LlAIH^
№
舄~。
兰击 肖 №D J>Nl少 OI^ 一j
,)
o
Me\/Me N
击一 ∞k叼竺·
图2方法1的合成路线
[Key words]dapoxetine;antidepressant;premature ejaculation;synthesis
达泊西汀(dapoxetine,LY-210448,1)是一个选 择性的5.羟色胺再摄取抑制剂,结构上类似于氟西 汀并同样具有抗抑郁作用,临床研究用其盐酸
万方数据
[17]
CROSS JH.Topiramate monotherapy for childhood absence sei‘ zures:an openlabel pilot study[J].Seizure,2002,11(6):406— 410.
[18]PRIVITERA MD,BRODIE MJ,MATTSON RH,et a1.Topira—
American Academy of Neurology and the American Epilepsy Soci。
ety[J].Neurology,2004,62(8):1252—1260.
编辑:杨青/接受日期:2006—09—10
达泊西汀的合成进展
张飞皇1,陈瞍2,徐云根1 (1中国药科大学新药研究中心,南京210009;2常州市新北区生产力促进中心,常州213022)
[19]
SILBERSTEIN SD,BEN—MENACHEM E,SHANK RP,et a1.
Topiramate monotherapy in epilepsy and migraine prevention[J].
Clin Ther,2005,27(2):154—165.
[20]
VALENCIA I,FONS C,KOTHARE SV,et a1.Efficacy and toler- ability of topiramate in children younger than 2 years old[J].J Child Neurof,2005,20(8):667—669.