la1007846_Hierarchical Carbon Foams with Independently Tunable Mesopore and Macropore Size Distribut
格力公司环保产品中有害物质控制管理规定
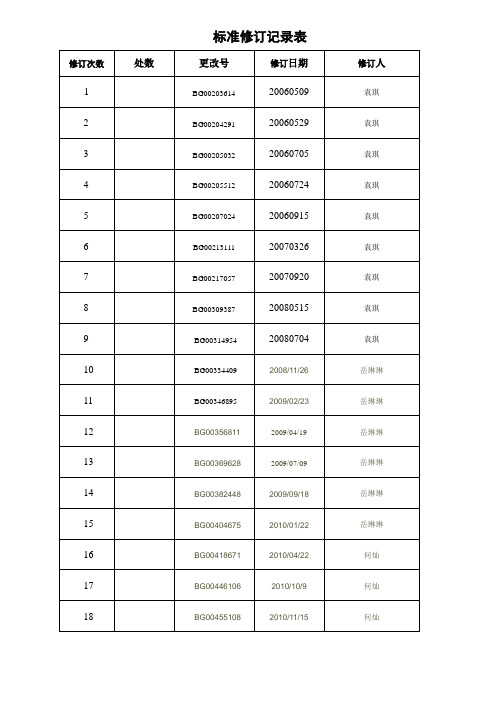
标准修订记录表QJ环保产品中有害物质控制管理规定珠海格力电器股份有限公司发布目次前言 (II)引言 (III)1 范围 (1)2 规范性引用文件 (1)3 术语和定义 (1)4 Ⅰ级有害物质具体管理标准 (5)5 Ⅱ级有害物质具体管理标准 (23)6 有害物质环保标识管理及对供应商的要求 (25)附录A(资料性附录)RoHS同质材料计算方法及部分环境管理物质的详细信息 (26)附录B(资料性附录) 各主要国家和地区就有害物质使用实施的法律法规 (35)附录C(资料性附录) 免于注册的物质清单 (39)附录D(资料性附录)注册需提交的信息 (40)附录E(资料性附录) 我司重点物料关注表 (42)附录F(规范性附录) REACH限制物质清单及使用要求 (43)附录G(资料性附录)第1批十五种高关注物质的检测流程 (59)参考文献 (62)前言格力电器股份有限公司技术标准是公司标准化委员会发布的标准,是公司内部使用的技术法规性文件。
本标准规定了格力电器产品需要满足的有害物质控制标准,检验分析方法等,旨在使原材料供应商和公司内部生产采用一致的控制标准,以符合欧盟及其他有关国家和地区或客户对电子电器产品中有毒有害物质的控制要求。
本标准与上次下发的版本相比的主要变化如下:--根据欧盟RoHS豁免条款修订决议修改本标准中关于RoHS各项豁免的表述(见4.1中表4,表6,表8,表9)本标准的附录A、附录B、附录C、附录D、附录F为资料性附录,附录E为规范性附录。
本标准由珠海格力电器股份有限公司提出。
本标准由珠海格力电器股份有限公司标准化委员会归口.本标准由珠海格力电器股份有限公司制冷技术研究院起草。
本标准主要起草人:王茜、袁琪、曾伟强、蔡小洪、颜小琳、龙新文、郑文力、古文育、唐君、高海涛、刘思红、张华、岳琳琳。
本标准于2005年12月首次发布,2006年5月第一次修订,2006年5月第二次修订,2006年7月第三次修订,2006年7月第四次修订,2006年9月第五次修订,2007年3月第六次修订,2007年9月第七次修订,2008年5月第八次修订,2008年7月第九次修订,2008年11月第十次修订,2009年2月第十一次修订,2009年4月第十二次修订,2009年7月第十三次修订,2009年9月第十四次修订,2010年1月第十五次修订,2010年4月第十六次修订,2010年10月第十七次修订,本次第十八次修订。
油漆名称中英文对照

油漆名称中英文对照Epoxy Zinc Rich Primer 环氧富锌漆MIO Epoxy Primer 环氧云铁漆Two‐pack Chlorinated Rubber Paint 2层氯化橡胶漆Acrylic Topcoat 丙烯酸面漆Incrganic Zinc Silicate Primer 无机硅酸锌底漆Modifed Polyester High‐temperature Paint 改性聚酯耐高温漆Heat resistant aluminium silicon paint 耐热铝硅漆Zinc silicate primer 硅酸锌底漆Epoxy Primer 环氧漆Polyurethane Paint 脂肪族聚氨酯面漆Silicone Aluminum 硅铝漆Penguard midcoat 环氧中间漆Epoxy mio barrio paint ,penguard Midcoat MIO 环氧云铁中间漆Hardtop AS Polyuerthane top 聚氨酯面漆Organic silicon high temperature tesistant paint 有机硅耐高温面漆Epoxy Primer(Zinc Free) 环氧底漆(无锌)Epoxy Mastic 环氧树脂乳香(RAL7035) Hempel 45880Epoxy Phenolic 环氧酚醛Hempel 8561Zinc Silicate Primer 硅酸锌Hempel 15700/1570GInorganic Silicate Topcoat 无机硅酸盐(珍珠灰)Amercoat 04Y31858A Epoxy Mastic 环氧树脂乳香Hempel 45880Zinc Rich Epoxy Primer 环氧富锌底漆Hempel 17360 Polyurethane Acrylic 聚氨酯丙烯酸(RAL7035) Hempel 55210 Polyamide cured epoxy primer 环氧聚酰胺底漆Polyamide cured epoxy(high build) 环氧聚酰胺底漆(高固性)Two‐pack high build aliphatic Polyurethan 2层高固性脂肪族聚氨酯漆(RAL9010) Inorgaic Zinc Silicate 无机硅酸锌(Gavosil 15700)Aluminium pigments of Silicone铝颜料硅树脂(Silicone Aluminium 56914) Polyamide cured epoxy primer环氧聚酰胺底漆Polyamide cured epoxy primer(high build)环氧聚酰胺底漆(高固性)Two‐pack high build aliphatic polyurethane 2层高固性脂肪族聚氨酯漆(RAL9010) Epoxy Zinc Rich Primer 环氧富锌漆MIO Epoxy Primer 环氧云铁漆Acrylic Topcoat 丙烯酸面漆(RAL9006)Epoxy Zinc Rich Primer 环氧富锌漆MIO Epoxy Primer 环氧云铁漆Two‐pack Chlorinated Rubber Paint 2层氯化橡胶漆(RAL9006)Inorgaic Zinc Silicate 无机硅酸锌Silicone Aluminium 硅胶铝Incrganic Zinc Silicate Primer 无机硅酸锌底漆Modifed Polyester High‐temperature Paint 改性聚酯耐高温漆Inorganic Zinc Silicate 无机硅酸锌Modifed Silicone Acrylics 改良有机硅丙烯漆zinc silicate primer 硅酸锌底漆Heat resistant aluminium silicon paint 耐热铝硅漆(RAL N7.0)Epoxy Zinc Rich Primer环氧富锌漆MIO Epoxy Primer 环氧云铁漆Futura AS,Acrylic Polyurethane top Coat 丙烯酸聚氨酯面漆(RAL7035)Zinc silicate primer 硅酸锌底漆Epoxy Primer 环氧漆Polyurethane Paint 脂肪族聚氨酯面漆 (RAL9010)Silicone aluminum 铝粉硅树脂Organic Primer 有机锌MIO Epoxy Primer 环氧漆Polyurethane Paint聚氨酯面漆(RAL 6024)Zinc silicate primer硅酸锌底漆Heat resistant aluminium silicon paint耐热铝硅漆(RAL N7.0)Epoxy phenolic resin paint环氧酚醛树脂漆Epoxy phenolic paint环氧酚醛漆Epoxy asphalt thick blade Primer环氧沥青厚桨底漆Epoxy asphalt thick blade Finish Coat(RAL 9005)环氧沥青厚桨底漆(RAL 9005) Epoxy Zinc Rich Primer 环氧富锌底漆Epoxy Polyamide paint环氧聚酰胺漆Two Pack Poly‐urethane Paint双组份聚亚安酯(RAL 1014)Poly‐urethane Paint聚亚安酯(RAL 1014)TwoPack Silicon acrylic Paint双组份硅丙烯酸(RAL 9006)Silicone heat-resistant aluminum paint 有机硅耐热铝漆High build epoxy intermediate paint 环氧厚浆中间漆。
好威皮具护理产品目录英文翻译

目录一步翻新色浆168A(油性).. 错误!未定义书签。
One-step refurbished mill base168 A (oil)一步翻新色浆168B(水性) .. 错误!未定义书签。
One-step refurbished mill base168 B(water)绒面磨砂翻新剂168C.............. 错误!未定义书签。
Suede nubuck refurbished agent 168C绒面皮改色剂............................. 错误!未定义书签。
Suede leather recolor agent绒面磨砂增艳剂......................... 错误!未定义书签。
Suede nubuck brightener agent红光去除剂................................. 错误!未定义书签。
Red dye remover环保超细无酪色膏(水性).......... 错误!未定义书签。
Environmental protection ultrafine no cream color paste (water)超艳荧光无酪色膏(水性).......... 错误!未定义书签。
Super bright fluorescent no cream color paste (water)金属颜料..................................... 错误!未定义书签。
Metallic pigment优丽高浓缩染料水..................... 错误!未定义书签。
Optimal high concentrated dye water皮革超能净面液......................... 错误!未定义书签。
Leather chaoneng net surface fluid强力去膜剂................................. 错误!未定义书签。
袄128小雯材料汇总

袄128小雯材料汇总袄128小雯是一种新型纤维素袄料。
具有优异的性能和广泛的适用性。
下面对袄128小雯的材料进行了详细介绍和分析,希望能够为读者提供有价值的信息。
一、原材料1、竹浆醋酸纤维竹浆醋酸纤维是袄128小雯的主要原材料之一,它是通过将竹浆用纯碱和醋酸进行酯化反应得到的。
竹浆醋酸纤维具有优异的抗菌、防臭、透气、吸湿、快干等特性,可以为袄128小雯的品质提供保障。
2、涤纶丝涤纶丝是一种合成纤维,它是袄128小雯的另一个原材料。
涤纶具有优异的强度、耐磨性、耐腐蚀性和易于染色等特点,可以为袄128小雯的外观和品质提供保证。
3、其他原材料除了竹浆醋酸纤维和涤纶丝,袄128小雯的生产还需要其他一些原材料,例如染料、酵素、纤维增性剂等。
这些原材料的选择和使用对袄128小雯的性能和质量都有着重要的影响。
二、生产工艺1、原料制备首先需要将涤纶丝和竹浆醋酸纤维进行拼混,以确保袄128小雯的透气性和吸湿性。
然后需要将拼混后的原料与染料、酵素、纤维增性剂等进行混合,以达到想要的颜色、手感、抗皱性等要求。
2、纺织加工在经过原料制备之后,就需要进行纺织加工了。
首先将原料分成纤维束,然后通过纺丝机将其纺织成纤维线。
接着,将纤维线通过织机进行织造,直到袄128小雯的成品形态出现。
3、后处理在经过纺织加工后,袄128小雯还需要进行后处理才能软化、定型和防皱。
后处理的过程包括洗涤、烘干、卷边、整烫等等。
这些后处理过程是非常重要的,可以保证袄128小雯的品质和性能符合标准要求。
三、应用领域1、服装袄128小雯的透气性和吸湿性非常好,适合用于制作夏季服装、内衣等。
而且它的手感柔软,质量轻,穿着舒适,是极受欢迎的面料之一。
2、家居用品袄128小雯的耐磨性强,易于清洁,具有抗菌防臭、透气吸湿等特性,适合用于制作各种家居用品,如窗帘、坐垫、桌布等等。
3、医疗卫生袄128小雯具有优异的抗菌、防臭、透气、吸湿等特性,适合用于制作医疗卫生用品,如口罩、披肩、手术衣等等。
科聚亚聚氨酯

HMDI耐黄变系统
品名 LW 520 LW 570 NCO % 4.75 7.5 硬度 90A 75D 固化剂种类 二胺类 二胺类 适用期(分钟) 4~6 1.5 特性 耐水解性佳 高定伸强度、耐水解性佳
6
LF-TDI聚醚低游离系统
品名 LF 800A LF 900A LF 930A LF 950A LF 600D LF 650D LF 700D LF 750D NCO % 2.9 3.8 5.05 6.05 7.25 7.7 8.25 8.9 硬度 80A 90A 93A 95A 60D 65D 70D 75D 固化剂种类 MOCA MOCA MOCA MOCA MOCA MOCA MOCA MOCA 适用期(分钟) 14 9 8.5 7.3 5.25 4.25 3.3 2.8 特性 易加工 动态性能佳、高回弹 动态性能佳、耐屈曲 高温动态性能佳 高温动态性能佳 高温动态性能佳 高温动态性能佳 高温动态性能佳
不断改善是科聚亚的使命。 致力于向客户提供更好的业务方案; 帮助员工学习与成长; 帮助投资者实现创新思考,稳固的发展规划以及满意结果。
1
科聚亚
聚氨酯业务
科聚亚聚氨酯在全球聚氨酯热浇注模塑预聚体、特殊水性聚氨酯分散体以及聚酯多元醇应用上处于领 先地位。我们的业务特性在于了解关注客户所需,提供适合的规格产品和应用开发以及完善的技术服 务与支持。 我们与客户的关系透过广泛全面的产品线和技术能力得以紧密联系,使得我们在业界以创造客户价值 的解决方案者著称。将您复杂而最具挑战的方案交给科聚亚,让我们和您一起开展双赢的方案。 科聚亚在全球提供超过300种聚氨酯预聚体和交联剂,包含Adiprene®, Vibrathane® 和Vibracure®等 高性能产品。 我们产品范围包含各种耐撕裂、耐磨浇注弹性体。它们在硬度、承载能力和耐刮、耐切上的杰出表现 在全球范围内赢得了客户的赞同。 科聚亚产品的主要应用领域包含: 工业和印刷滚轮; 矿山,原油和天然气田开采设备; 机械设备零件; 工业轮; 电器零件和输送设备; 各式运动器械和耗材。 除了传统的MDI、TDI预聚体,科聚亚也提供各种 TDI、 MDI、 PPDI和HDI种类低游离异氰酸酯预聚 体。这些低游离预聚体除了提供更好的物性,操作便利外,在确保环境安全和健康卫生方面也有杰出 的表现。我们的产品系列包括聚酯、聚醚、聚己内酰酯和聚碳酸酯类。 科聚亚提供Fomrez®聚酯多元醇应用于多种软质泡沫、热塑性弹性体、涂层、黏合剂和其它弹性体应 用。Witcobond®聚氨酯分散体具有优异的涂布性和接着性,广泛应用于木材、塑料、玻纤、化纤、皮 革、橡胶和其它多种材料。 商品名: 浇注型聚氨酯: Adiprene®和Vibrathane® 水性聚氨酯: Witcobond® 聚酯多元醇: Fomrez® 高硬度塑料系统: Royalcast®
华奇(中国)化工有限公司

华奇(中国)化工有限公司
佚名
【期刊名称】《橡胶科技》
【年(卷),期】2015(000)001
【摘要】<正>责任严谨创新全球大型橡胶及轮胎行业用酚醛树脂及间苯二酚树脂厂商主要产品"对-特辛基苯酚甲醛增粘树脂"SL-1800系列主要牌号有:SL-1801,SL-1802,SL-1801LFP(低游离酚增粘树脂),SL-1805。
SL-1800系列产品是非热反应性的热塑性树脂,具有优异的增粘性能,可用于任何需要高粘度粘接的复合橡胶制品,尤其是合成橡胶制品。
"对-叔丁基酚醛增粘树脂"SL-1400系列主要牌号有:SL-1401,SL-1402,SL-1403,SL-1405,SL-1407,SL-1408,SL-T421。
【总页数】1页(P2)
【正文语种】中文
【中图分类】F416.72
【相关文献】
1.华奇(中国)化工有限公司 [J], ;
2.华奇(中国)化工有限公司 [J], ;
3.华奇(张家港)化工有限公司 [J], ;
4.华奇(中国)化工有限公司 [J], ;
5.智能、开放、融合,铸就研华新格局——访研华(中国)有限公司工业自动化事业群中国区总经理蔡奇男先生 [J],
因版权原因,仅展示原文概要,查看原文内容请购买。
标准红外光谱图谱

Go to: home • ir • proton nmr • carbon nmr• mass specTable of Contents - IRI. HydrocarbonsII. Halogenated HydrocarbonsIII. Nitrogen Containing CompoundsIV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O And P(=O)-O) VI. Sulfur Containing CompoundsVII. Oxygen Containing Compounds (Except -C(=O)-)VIII. Compounds Containing Carbon To Oxygen Double BondsI. HydrocarbonsA. Saturated Hydrocarbons1. Normal Alkanes2. Branched Alkanes3. Cyclic AlkanesB. Unsaturated Hydrocarbons1. Acyclic Alkenes2. Cyclic Alkenes3. AlkynesC. Aromatic Hydrocarbons1. Monocyclic (Benzenes)2. PolycyclicII. Halogenated HydrocarbonsA. Fluorinated Hydrocarbons1. Aliphatic2. AromaticB. Chlorinated Hydrocarbons1. Aliphatic2. Olefinic3. AromaticC. Brominated Hydrocarbons1. Aliphatic2. Olefinic3. AromaticD. Iodinated Hydrocarbons1. Aliphatic and Olefinic2. AromaticIII. Nitrogen Containing CompoundsA. Amines1. Primarya. Aliphatic and Olefinicb. Aromatic2. Secondarya. Aliphatic and Olefinicb. Aromatic3. Tertiarya. Aliphatic and Olefinicb. AromaticB. PyridinesC. QuinolinesD. Miscellaneous Nitrogen HeteroaromaticsE. HydrazinesF. Amine SaltsG. Oximes (-CH=N-OH)H. Hydrazones (-CH=N-NH2)I. Azines (-CH=N-N=CH-)J. Amidines (-N=CH-N)K. Hydroxamic AcidsL. Azo Compounds (-N=N-)M. Triazenes (-N=N-NH-)N. Isocyanates (-N=C=O)O. Carbodiimides (-N=C=N-)P. Isothiocyanates (-N=C=S)Q. Nitriles (-C≡N)1. Aliphatic2. Olefinic3. AromaticR. Cyanamides (=N-C≡N)S. Thiocyanates (-S-C≡N)T. Nitroso Compounds (-N=O)U. N-Nitroso Compounds (=N-N=O)V. Nitrites (-O-N=O)W. Nitro Compounds (-NO2)1. Aliphatic2. AromaticX. N-Nitro-Compounds (=N-NO2)IV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O and P(=O)-O) VI. Sulfur Containing CompoundsA. Sulfides (R-S-R)1. Aliphatic2. Heterocyclic3. AromaticB. Disulfides (R-S-S-R)C. Thiols1. Aliphatic2. AromaticD. Sulfoxides (R-S(=O)-R)E. Sulfones (R-SO2-R)F. Sulfonyl Halides (R-SO2-X)G. Sulfonic Acids (R-SO2-OH)1. Sulfonic Acid Salts (R-SO2-O-M)2. Sulfonic Acid Esters (R-SO2-O-R)3. Sulfuric Acid Esters (R-O-S(=O)-O-R)H. Thioamides (R-C(=S)-NH2)I. Thioureas (R-NH-C(=S)-NH2)J. Sulfonamides (R-SO2-NH2)K. Sulfamides (R-NH-SO2-NH-R)VII. Oxygen Containing Compounds (Except -C(=O)-)A. Ethers1. Aliphatic Ethers (R-O-R)2. Acetals (R-CH-(-O-R)2)3. Alicyclic Ethers4. Aromatic Ethers5. Furans6. Silicon Ethers (R3-Si-O-R)7. Phosphorus Ethers ((R-O)3-P)8. Peroxides (R-O-O-R)B. Alcohols (R-OH)1. Primarya. Aliphatic and Alicyclicb. Olefinicc. Aromaticd. Heterocyclic2. Secondarya. Aliphatic and Alicyclicb. Olefinicc. Aromatic3. Tertiarya. Aliphaticb. Olefinicc. Aromatic4. Diols5. Carbohydrates6. PhenolsVIII. Compounds Containing Carbon To Oxygen Double BondsA. Ketones (R-C(=O)-R)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. α-Diketones and β-DiketonesB. Aldehydes (R-C(=O)-H)C. Acid Halides (R-C(=O)-X)D. Anhydrides (R-C(=O)-O-C(=O)-R)E. Amides1. Primary (R-C(=O)-NH2)2. Secondary (R-C(=O)-NH-R)3. Tertiary (R-C(=O)-N-R2)F. Imides (R-C(=O)-NH-C(=O)-R)G. Hydrazides (R-C(=O)-NH-NH2)H. Ureas (R-NH-C(=O)-NH2)I. Hydantoins, Uracils, BarbituratesJ. Carboxylic Acids (R-C(=O)-OH)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. Amino Acids5. Salts of Carboxylic AcidsK. Esters1. Aliphatic Esters of Aliphatic Acids2. Olefinic Esters of Aliphatic Acids3. Aliphatic Esters of Olefinic Acids4. Aromatic Esters of Aliphatic Acids5. Esters of Aromatic Acids6. Cyclic Esters (Lactones)7. Chloroformates8. Esters of Thio-Acids9. Carbamates10. Esters of Phosphorus AcidsPublished by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specTable of Contents - Proton NMRI. HydrocarbonsII. Halogenated HydrocarbonsIII. Nitrogen Containing CompoundsIV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O and P(=O)-O) VI. Sulfur Containing CompoundsVII. Oxygen Containing Compounds (Except -C(=O)-)VIII. Compounds Containing Carbon To Oxygen Double BondsI. HydrocarbonsA. Saturated Hydrocarbons1. Normal Alkanes2. Branched Alkanes3. Cyclic AlkanesB. Unsaturated Hydrocarbons1. Acyclic Alkenes2. Cyclic Alkenes3. AlkynesC. Aromatic Hydrocarbons1. Monocyclic (Benzenes)2. PolycyclicII. Halogenated HydrocarbonsA. Fluorinated Hydrocarbons1. Aliphatic2. AromaticB. Chlorinated Hydrocarbons1. Aliphatic2. AromaticC. Brominated Hydrocarbons1. Aliphatic2. AromaticD. Iodinated Hydrocarbons1. Aliphatic2. AromaticIII. Nitrogen Containing CompoundsA. Amines1. Primarya. Aliphaticb. Aromatic2. Secondarya. Aliphaticb. Aromatic3. Tertiarya. Aliphaticb. AromaticB. PyridinesC. Quaternary Ammonium SaltsD. HydrazinesE. Amine SaltsF. Ylidene Compounds (-CH=N-)G. Oximes (-CH=N-OH)H. Hydrazones (-CH=N-NH2)I. Azines (-CH=N-N=CH-)J. Amidines (-N=CH-N)K. Hydroxamic AcidsL. Azo Compounds (-N=N-)M. Isocyanates (-N=C=O)N. Carbodiimides (-N=C=N-)O. Isothiocyanates (-N=C=S)P. Nitriles (-C≡N)1. Aliphatic2. Olefinic3. AromaticQ. Cyanamides (=N-C≡N)R. Isocyanides (-N≡C )S. Thiocyanates (-S-C≡N)T. Nitroso Compounds (-N=O)U. N-Nitroso Compounds (=N-N=O)V. Nitrates (-O-NO2)W. Nitrites (-O-N=O)X. Nitro Compounds (-NO2)1. Aliphatic2. AromaticY. N-Nitro-Compounds (=N-NO2)IV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O and P(=O)-O) VI. Sulfur Containing CompoundsA. Sulfides (R-S-R)1. Aliphatic2. AromaticB. Disulfides (R-S-S-R)C. Thiols1. Aliphatic2. AromaticD. Sulfoxides (R-S(=O)-R)E. Sulfones (R-SO2-R)F. Sulfonyl Halides (R-SO2-X)G. Sulfonic Acids (R-SO2-OH)1. Sulfonic Acid Salts (R-SO2-O-M)2. Sulfonic Acid Esters (R-SO2-O-R)3. Sulfuric Acid Esters (R-O-S(=O)-O-R)4. Sulfuric Acid Salts (R-O-SO2-O-M)H. Thioamides (R-C(=S)-NH2)I. Thioureas (R-NH-C(=S)-NH2)J. Sulfonamides (R-SO2-NH2)VII. Oxygen Containing Compounds (Except -C(=O)-)A. Ethers1. Aliphatic Ethers (R-O-R)2. Alicyclic Ethers3. Aromatic Ethers4. Furans5. Silicon Ethers (R3-Si-O-R)6. Phosphorus Ethers ((R-O)3-P)B. Alcohols (R-OH)1. Primarya. Aliphaticb. Olefinicc. Aromatic2. Secondarya. Aliphaticb. Aromatic3. Tertiarya. Aliphaticb. Aromatic4. Diols and Polyols5. Carbohydrates6. PhenolsVIII. Compounds Containing Carbon To Oxygen Double BondsA. Ketones (R-C(=O)-R)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. a-Diketones and b-DiketonesB. Aldehydes (R-C(=O)-H)C. Acid Halides (R-C(=O)-X)D. Anhydrides (R-C(=O)-O-C(=O)-R)E. Amides1. Primary (R-C(=O)-NH2)2. Secondary (R-C(=O)-NH-R)3. Tertiary (R-C(=O)-N-R2)F. Imides (R-C(=O)-NH-C(=O)-R)G. Hydrazides (R-C(=O)-NH-NH2)H. Ureas (R-NH-C(=O)-NH2)I. Hydantoins, Uracils, BarbituratesJ. Carboxylic Acids (R-C(=O)-OH)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. Amino Acids5. Salts of Carboxylic AcidsK. Esters1. Aliphatic Esters of Aliphatic Acids2. Olefinic Esters of Aliphatic Acids3. Aromatic Esters of Aliphatic Acids4. Cyclic Esters (Lactones)5. Chloroformates6. Carbamates7. Esters of Phosphorus AcidsPublished by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specTable of Contents - Carbon NMRI. HydrocarbonsII. Halogenated HydrocarbonsIII. Nitrogen Containing CompoundsIV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O And P(=O)-O) VI. Sulfur Containing CompoundsVII. Oxygen Containing Compounds (Except -C(=O)-)VIII. Compounds Containing Carbon To Oxygen Double BondsI. HydrocarbonsA. Saturated Hydrocarbons1. Normal Alkanes2. Branched Alkanes3. Cyclic AlkanesB. Unsaturated Hydrocarbons1. Acyclic Alkenes2. AlkynesC. Aromatic Hydrocarbons1. Monocyclic (Benzenes) and PolycyclicII. Halogenated HydrocarbonsA. Fluorinated Hydrocarbons1. Aliphatic2. AromaticB. Chlorinated Hydrocarbons1. Aliphatic2. AromaticC. Brominated Hydrocarbons1. Aliphatic2. AromaticD. Iodinated Hydrocarbons1. Aliphatic2. AromaticIII. Nitrogen Containing CompoundsA. Amines1. Primarya. Aliphaticb. Aromatic2. Secondarya. Aliphaticb. Aromatic3. Tertiarya. Aliphaticb. AromaticB. PyridinesC. Amine SaltsD. Oximes (-CH=N-OH)E. Quaternary Ammonium SaltsF. Nitriles (-C≡N)1. Aliphatic2. Olefinic3. AromaticG. Thiocyanates (-S-C≡N)H. Nitro Compounds (-NO2)1. Aliphatic2. AromaticIV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O and P(=O)-O) VI. Sulfur Containing CompoundsA. Sulfides (R-S-R)1. Aliphatic2. AromaticB. Disulfides (R-S-S-R)C. Thiols1. Aliphatic2. AromaticD. Sulfones (R-SO2-R)VII. Oxygen Containing Compounds (Except -C(=O)-)A. Ethers1. Aliphatic Ethers (R-O-R)2. Alicyclic Ethers3. Aromatic EthersB. Alcohols (R-OH)1. Primarya. Aliphatic and Alicyclicb. Aromatic2. Secondarya. Aliphatic and Alicyclic3. Tertiarya. Aliphatic4. PhenolsVIII. Compounds Containing Carbon To Oxygen Double BondsA. Ketones (R-C(=O)-R)1. Aliphatic and Alicyclic2. AromaticB. Aldehydes (R-C(=O)-H)C. Acid Halides (R-C(=O)-X)D. Anhydrides (R-C(=O)-O-C(=O)-R)E. Amides1. Primary (R-C(=O)-NH2)2. Secondary (R-C(=O)-NH-R)3. Tertiary (R-C(=O)-N-R2)F. Carboxylic Acids (R-C(=O)-OH)1. Aliphatic and Alicyclic2. AromaticG. Esters1. Aliphatic Esters of Aliphatic Acids2. Olefinic Esters of Aliphatic Acids3. Aromatic Esters of Aliphatic AcidsPublished by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specTable of Contents - MSComing SoonI. HydrocarbonsII. Halogenated HydrocarbonsIII. Nitrogen Containing CompoundsIV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O And P(=O)-O) VI. Sulfur Containing CompoundsVII. Oxygen Containing Compounds (Except -C(=O)-)VIII. Compounds Containing Carbon To Oxygen Double BondsI. HydrocarbonsA. Saturated Hydrocarbons1. Normal Alkanes2. Branched Alkanes3. Cyclic AlkanesB. Unsaturated Hydrocarbons1. Acyclic Alkenes2. Cyclic Alkenes3. AlkynesC. Aromatic Hydrocarbons1. Monocyclic (Benzenes)2. PolycyclicII. Halogenated HydrocarbonsA. Fluorinated Hydrocarbons1. Aliphatic2. AromaticB. Chlorinated Hydrocarbons1. Aliphatic2. Olefinic3. AromaticC. Brominated Hydrocarbons1. Aliphatic2. Olefinic3. AromaticD. Iodinated Hydrocarbons1. Aliphatic and Olefinic2. AromaticIII. Nitrogen Containing CompoundsA. Amines1. Primarya. Aliphatic and Olefinicb. Aromatic2. Secondarya. Aliphatic and Olefinicb. Aromatic3. Tertiarya. Aliphatic and Olefinicb. AromaticB. PyridinesC. QuinolinesD. Miscellaneous Nitrogen HeteroaromaticsE. HydrazinesF. Amine SaltsG. Oximes (-CH=N-OH)H. Hydrazones (-CH=N-NH2)I. Azines (-CH=N-N=CH-)J. Amidines (-N=CH-N)K. Hydroxamic AcidsL. Azo Compounds (-N=N-)M. Triazenes (-N=N-NH-)N. Isocyanates (-N=C=O)O. Carbodiimides (-N=C=N-)P. Isothiocyanates (-N=C=S)Q. Nitriles (-C≡N)1. Aliphatic2. Olefinic3. AromaticR. Cyanamides (=N-C≡N)S. Thiocyanates (-S-C≡N)T. Nitroso Compounds (-N=O)U. N-Nitroso Compounds (=N-N=O)V. Nitrites (-O-N=O)W. Nitro Compounds (-NO2)1. Aliphatic2. AromaticX. N-Nitro-Compounds (=N-NO2)IV. Silicon Containing Compounds (Except Si-O)V. Phosphorus Containing Compounds (Except P-O and P(=O)-O) VI. Sulfur Containing CompoundsA. Sulfides (R-S-R)1. Aliphatic2. Heterocyclic3. AromaticB. Disulfides (R-S-S-R)C. Thiols1. Aliphatic2. AromaticD. Sulfoxides (R-S(=O)-R)E. Sulfones (R-SO2-R)F. Sulfonyl Halides (R-SO2-X)G. Sulfonic Acids (R-SO2-OH)1. Sulfonic Acid Salts (R-SO2-O-M)2. Sulfonic Acid Esters (R-SO2-O-R)3. Sulfuric Acid Esters (R-O-S(=O)-O-R)H. Thioamides (R-C(=S)-NH2)I. Thioureas (R-NH-C(=S)-NH2)J. Sulfonamides (R-SO2-NH2)K. Sulfamides (R-NH-SO2-NH-R)VII. Oxygen Containing Compounds (Except -C(=O)-)A. Ethers1. Aliphatic Ethers (R-O-R)2. Acetals (R-CH-(-O-R)2)3. Alicyclic Ethers4. Aromatic Ethers5. Furans6. Silicon Ethers (R3-Si-O-R)7. Phosphorus Ethers ((R-O)3-P)8. Peroxides (R-O-O-R)B. Alcohols (R-OH)1. Primarya. Aliphatic and Alicyclicb. Olefinicc. Aromaticd. Heterocyclic2. Secondarya. Aliphatic and Alicyclicb. Olefinicc. Aromatic3. Tertiarya. Aliphaticb. Olefinicc. Aromatic4. Diols5. Carbohydrates6. PhenolsVIII. Compounds Containing Carbon To Oxygen Double BondsA. Ketones (R-C(=O)-R)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. α-Diketones and β-DiketonesB. Aldehydes (R-C(=O)-H)C. Acid Halides (R-C(=O)-X)D. Anhydrides (R-C(=O)-O-C(=O)-R)E. Amides1. Primary (R-C(=O)-NH2)2. Secondary (R-C(=O)-NH-R)3. Tertiary (R-C(=O)-N-R2)F. Imides (R-C(=O)-NH-C(=O)-R)G. Hydrazides (R-C(=O)-NH-NH2)H. Ureas (R-NH-C(=O)-NH2)I. Hydantoins, Uracils, BarbituratesJ. Carboxylic Acids (R-C(=O)-OH)1. Aliphatic and Alicyclic2. Olefinic3. Aromatic4. Amino Acids5. Salts of Carboxylic AcidsK. Esters1. Aliphatic Esters of Aliphatic Acids2. Olefinic Esters of Aliphatic Acids3. Aliphatic Esters of Olefinic Acids4. Aromatic Esters of Aliphatic Acids5. Esters of Aromatic Acids6. Cyclic Esters (Lactones)7. Chloroformates8. Esters of Thio-Acids9. Carbamates10. Esters of Phosphorus AcidsPublished by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specSaturated HydrocarbonsNormal Alkanes1. C-H stretching vibration:CH3 asymmetric stretching, 2972-2952 cm-1CH3 symmetric stretching, 2882-2862 cm-1CH2 asymmetric stretching, 2936-2916 cm-1CH2 symmetric stretching, 2863-2843 cm-12. C-H bending vibration:CH3 asymmetric bending, 1470-1430 cm-1CH2 asymmetric bending, 1485-1445 cm-1(overlaps band due to CH3 asymmetricbending)3. C-H bending vibration:CH3 symmetric bending, 1380-1365 cm-1(when CH3 is attached to a C atom)4. C-H wagging vibration:CH2 out-of-plane deformations wagging, 1307-1303 cm-1 (weak) 5. CH2 rocking vibration:(CH2)2 in-plane deformations rocking, 750-740 cm-1(CH2)3 in-plane deformations rocking, 740-730 cm-1(CH2)4 in-plane deformations rocking, 730-725 cm-1(CH2) ≥ 6 in-plane deformations rocking, 722 cm-1Splitting of the absorption band occurs in most cases (730 and 720 cm-1) when the long carbon-chain alkane is in the crystalline state (orthorombic or monoclinic form).Coming Soon!Click on a vibrational mode link in the table to the leftor the spectrum above to visualize the vibrational mode here.Published by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Saturated HydrocarbonsBranched Alkanes1. C-H stretching vibration:CH3 asymmetric stretching, 2972-2952 cm-1CH3 symmetric stretching, 2882-2862 cm-1CH2 asymmetric stretching, 2936-2916 cm-1CH2 symmetric stretching, 2863-2843 cm-12. C-H bending vibration:CH3 asymmetric bending, 1470-1430 cm-1CH2 asymmetric bending, 1485-1445 cm-1(overlaps band due to CH3 symmetric bending)3. C-H bending vibration:-C-C(CH3)-C-C- symmetric bending, 1380-1365 cm-1(when CH3 is attached to a C atom)-C-C(CH3)-C(CH3)-C-C- symmetric bending, 1380-1365 cm-1(when CH3 is attached to a C atom)(CH3)2CH- symmetric bending, 1385-1380 cm-1and 1365 cm-1(two bands of about equal intensity)-C-C(CH3)2-C- symmetric bending,1385-1380 cm-1and 1365 cm-1 (two bands of about equal intensity).(CH3)3C- symmetric bending, 1395-1385 cm-1and 1365 cm-1(two bands of unequal intensity with the 1365 cm-1 band as the much stronger component of the doublet).4. Skeletal vibration:-C-C(CH3)-C-C-,1159-1151cm-1-C-C(CH3)-C(CH3)-C-C-,1130-1116 cm-1(CH3)CH-,1175-1165 cm-1 and 1170-1140 cm-1-C-C(CH3)2-C-,1192-1185 cm-1(CH3)3C-, 1255-1245 cm-1 and 1250-1200 cm-15. C-H rocking vibration:(CH2)2 in-plane deformations rocking, 750-740 cm-1(CH2)3 in-plane deformations rocking, 740-730 cm-1(CH2)4 in-plane deformations rocking, 730-725 cm-1(CH2) ≥ 6 in-plane deformations rocking, 722 cm-1Coming Soon!Click on a vibrational mode link in the table to the left or the spectrum above to visualize the vibrational modehere.Published by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Saturated Hydrocarbons Cyclic AlkanesCyclopropanes1. C-H stretching vibration:ring CH 2 asymmetric stretching, 3100-3072 cm -1 ring CH 2 symmetric stretching, 3030-2995 cm -12. Ring deformation vibration:ring deformation, 1050-1000 cm -13. C-H deformation vibration: CH 2 wagging, 860-790 cm -1Cyclobutanes1. C-H stretching vibration:ring CH 2 asymmetric stretching, 3000-2974 cm -1 ring CH 2 symmetric stretching, 2925-2875 cm -12. C-H deformation vibration:ring CH 2 asymmetric bending, ca 1444 cm -13. Ring deformation vibration:ring deformation, 1000-960 cm -1 888-838 cm -14. C-H deformation vibration:ring CH 2 rocking, 950-900 cm -1Cyclopentanes1. C-H stretching vibration:ring CH 2 asymmetric stretching, 2960-2952 cm -1 ring CH 2 symmetric stretching, 2866-2853 cm -1 2. C-H deformation vibration:ring CH 2 asymmetric bending, ca 1455 cm -1 3. Ring deformation vibration:ring deformation, 1000-960 cm -1 4. C-H deformation vibration:ring CH 2rocking, 930-890 cm -1Cyclohexanes1. C-H stretching vibration:ring CH 2 asymmetric stretching, ca 2927 cm -1ring CH 2 symmetric stretching, ca 2854 cm -1 2. C-H deformation vibration:ring CH 2 asymmetric bending, ca 1462 cm -1 3. C-H deformation vibration:ring CH 2 wagging, ca 1260 cm -1 4. Ring deformation vibration:ring deformation, 1055-1000 cm -1 1000- 952 cm -1 5. C-H deformation vibration:ring CH 2 rocking, 890-860 cm -16. The spectra of cyclic alkanes of five or more ring carbons show ring CH 2 stretching frequencies which overlap those of CH 3 and CH 2 groups of their alkyl substituents. These frequencies also overlap thoseof the CH 3 and CH 2 stretching frequencies of acylic alkanes. When samples of unknown composition are examined for the presence of such ring structures, the absorption bands of their spectra at the C-H stretching region should havethe best possible resolution.Coming Soon!Click on a vibrational mode link in the table to the left or the spectrum above to visualize the vibrational modehere.Numerous references cite the spectral region of 2800-2600 cm-1 for obtainingconfirmatory evidence of the presence of saturated simple ring structures. Absorptionat this region consists of a weak band or bands whose pattern and band locations arehelpful in confirming or indicating the presence of these rings. Although such absorptionfeatures have a limited diagnostic value, it is most reliable when the absorption occursin the spectra of simple saturated aliphatic hydrocarbons.Cycloalkanes (8, 9, and 10 C atoms)1 C-H stretching vibration:ring CH2 asymmetric stretching, ca 2930 cm-1ring CH2 symmetric stretching, ca 2850 cm-12. C-H deformation vibration:ring CH2 asymmetric bending, 2 or 3 absorption bands,1487-1443 cm-1Published by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specUnsaturated HydrocarbonsAcyclic AlkenesMonosubstituted Alkenes (vinyl)1. C=C stretching vibration:C=C stretching, 1648-1638 cm-12. C-H deformation vibration:trans CH wagging, 995-985 cm-1CH2 wagging, 910-905 cm-13. C-H stretching vibration:CH2 asymmetric stretching, 3092-3077 cm-1CH2 symmetric stretching and CH stretching, 3025-3012 cm-1 4. C-H deformation vibration:CH2 asymmetric bending, 1420-1412 cm-15. C-H deformation vibration overtone:overtone of CH2 wagging, 1840-1805 cm-1Asymmetric Disubstituted Alkenes (vinylidine)1. C=C stretching vibration:C=C stretching, 1661-1639 cm-12. C-H deformation vibration:CH2 wagging, 895-885 cm-13. C-H stretching vibration:CH2 stretching asymmetric, 3100-3077 cm-14. C-H deformation vibration overtone:overtone of CH2 wagging, 1792- 1775 cm-1Symmetric Disubstituted Alkenes (cis)1. C=C stretching vibration:C=C stretching, 1662- 1631 cm-12. C-H deformation vibration:cis CH wagging, 730- 650 cm-13. C-H stretching vibration:CH stretching, 3050-3000 cm-1Symmetric Disubstituted Alkenes (trans)1. C=C stretching vibration:C=C stretching, ca 1673 cm-1, very weak or absent2. C-H deformation vibration:trans CH wagging, 980-965 cm-13. C-H stretching vibration:CH stretching, 3050-3000 cm-1Trisubstituted Alkenes1. C=C stretching vibration:C=C stretching, 1692-1667 cm-12. C—H deformation vibration:C-H wagging, 840-790 cm-13. C-H stretching vibration:C-H stretching, 3050-2990 cm-1Coming Soon!Click on a vibrational mode link in the table to the left or the spectrum above to visualize the vibrational modehere.Tetrasubstituted Alkenes1. C=C stretching vibration:C=C stretching, 1680-1665 cm-1, very weak or absentNOTES: The C=C stretching vibration of molecules which maintain acenter of symmetry absorbs very weakly, if at all, in the infrared region and,usually, is difficult to detect. This is true of the trans isomers and thetetrasubstitutedC=C linkages.When two or more olefinic groups occur in the hydrocarbon molecule, the infraredabsorption spectrum shows the additive and combined absorption of theunsaturatedgroups. However, if the unsaturated groups are subject to conjugation, the C=Cstretchingfrequency, usually, is lowered and a splitting of the C=C stretching frequencyband occurs.Conjugation also intensifies the C=C stretching frequency of trans unsaturatedgroups.Published by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Go to: home • ir • proton nmr • carbon nmr• mass specUnsaturated Hydrocarbons Cyclic AlkenesEndocyclic C=CEndocyclic C=C corresponds to cis symmetrically disubstituted C=C of acyclic alkenes.1. C=C stretching, vibration:C=C stretching, near 1650 cm -1(except cyclobutene, 1560 cm -1 and cyclopentene, 1611 cm -1)2. C-H deformation vibration: CH wagging, 730- 650 cm -13. C-H stretching vibration:CH stretching, 3075- 3010 cm -1(usually two bands, asymmetric stretching and symmetric stretching for 4, 6, 7, and 8 membered rings)1- substituted endocyclic C=C1- substituted endocyclic C=C corresponds to trisubstituted acyclic alkenes.1. C=C stretching vibration:C=C stretching, near 1650 cm -1 (frequency raised)2. C-H deformation vibration: CH wagging, 840-790 cm -13. C-H stretching vibration:CH stretching, near 3000 cm -11.2- disubstituted endocyclic C=C1. C=C stretching vibration:C=C stretching, 1690-1670 cm -1 (4, 5, and 6 membered rings)Exocyclic C=CH 2Exocyclic C=CH 2 corresponds to the asymmetrically disubstituted C=C of acyclic alkenes (vinylidine).1. C=C stretching,1678-1650 cm -1 (4, 5, and 6 membered rings)2. C-H deformation vibration:=CH 2 wagging, 895-885 cm -13. C-H stretching vibration:=CH 2 stretching, near 3050 cm -1NOTES: The C=C stretching frequency of both the endocyclic HC=CH and the exocyclic C=CH 2 is sensitive to ring strain. As the ring size decreases from 6 to 4 members, the C=C stretching frequency of the endocyclic HC=CH is lowered. However, for the C=C stretching frequency of exocyclic C=CH 2, a gradual increase in the C=C stretching frequency occurs as the ring gets smaller. Substitution of methyl groups for the hydrogens of the endocyclic HC=CH and the exocyclic C=CH 2 cause an increase in the C=C stretching frequency.When two or more C=C groups occur in the hydrocarbon molecule, the infrared absorption spectrum shows the additive and combined absorption effects of the unsaturated groups. If such groups are subject to conjugation, the C=C stretching frequency is lowered and asplitting of the C=C stretching frequency band occurs.Coming Soon!Click on a vibrational mode link in the table to the left or the spectrum above to visualize the vibrational modehere.Published by Bio-Rad Laboratories, Inc., Informatics Division. © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.Unsaturated Hydrocarbons AlkynesMonosubstituted Alkynes (RC ≡CH)1. C ≡C stretching vibration:C ≡C stretching, 2140-2100 cm -12. C-H stretching vibration:≡CH bending, ca 3300 cm -13. C-H deformation vibration: ≡CH bending, 642-615 cm -14. C-H deformation vibration overtone:overtone of ≡CH deformation, 1260-1245 cm -1Disubstituted Alkynes (RC ≡CR')1. C ≡C stretching vibration:C ≡C stretching, 2260-2190 cm -1 (unconjugated)NOTES: Although the intensity of the absorption band caused bythe C ≡C stretching vibration is variable, it is strongest when the alkyne group is monosubstituted. When this group is disubstituted in open chain compounds, the intensity of the C ≡C stretching vibration band diminishesas its position in the molecule tends to establish a pseudo center of symmetry. In some instances this band is too weak to be detected and, thus, its absence in the spectrum does not, necessarily, establish proof of the absence of this linkage.Occasionally, the spectra of disubstituted alkynes show two or more bands at the C ≡C stretching region.Conjugation with olefinic double bonds or aromatic rings tend to slightly increase the intensity of the C ≡C stretching vibration band and shift it toa lower frequency.Coming Soon!Click on a vibrational mode link in the table to the left or the spectrum above to visualize the vibrational modehere.Published by Bio-Rad Laboratories, Inc., Informatics Division . © 1978-2004 Bio-Rad Laboratories, Inc. All Rights Reserved.。
汉高产品表
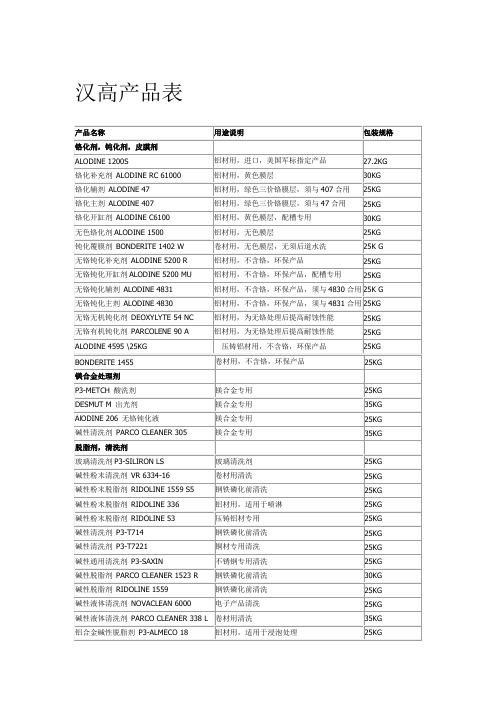
铝材用,不含铬,环保产品,须与 4830 合用 25K G 铝材用,不含铬,环保产品,须与 4831 合用 25KG 铝材用,为无铬处理后提高耐蚀性能 铝材用,为无铬处理后提高耐蚀性能 压铸铝材用,不含铬,环保产品 卷材用,不含铬,环保产品 25KG 25KG 25KG 25KG
碱性液体清洗剂 PARCO CLEANER 338 L 卷材用清洗 铝合金碱性脱脂剂 P3-ALMECO 18 铝材用,适用于浸泡处理
30KG 25KG 30KG 35KG 25KG 25KG 25KG
铜拉丝专用
180KG 20KG 25KG 30KG 25KG
铝碱蚀亚光添加剂
30KG 25KG 20KG
专用于有机颜料染色后高温封闭
25KG 25KG
25KG 25KG 25K G 25KG 25KG 25KG 25KG 25KG 35KG 35KG 25KG 35KG 35KG 25KG 25KG 25KG 25KG
压铸铝材用,环保产品
25KG
锌钙系磷化 锰系磷化前表调 锰系厚膜磷化 锌系低温磷化配槽剂 锌系低温磷化添加剂 喷淋法低温三元磷化配槽剂 喷淋法低温三元磷化添加剂 三元磷化配槽剂 三元磷化添加剂 浸渍法低温三元磷化配槽剂 浸渍法低温三元磷化添加剂 喷淋法三元磷化配槽剂 喷淋法三元磷化添加剂 常温磷化 无磷环保磷化 可同时磷化铁铝锌等 磷化同时可除油 磷化同时可除油 与 NT1 配套 磷化配套 磷化配套
汉高产品表
产品名称 铬化剂,钝化剂,皮膜剂 ALODINE 1200S 铬化补充剂 ALODINE RC 61000 铬化辅剂 ALODINE 47 铬化主剂 ALODINE 407 铬化开缸剂 ALODINE C6100 无色铬化剂 ALODINE 1500 钝化覆膜剂 BONDERITE 1402 W 无铬钝化补充剂 ALODINE 5200 R 无铬钝化开缸剂 ALODINE 5200 MU 无铬钝化辅剂 ALODINE 4831 无铬钝化主剂 ALODINE 4830 无铬无机钝化剂 DEOXYLYTE 54 NC 无铬有机钝化剂 PARCOLENE 90 A ALODINE 4595 \25KG BONDERITE 1455 镁合金处理剂 P3-METCH 酸洗剂 DESMUT M 出光剂 AlODINE 206 无铬钝化液 碱性清洗剂 PARCO CLEANER 305 脱脂剂,清洗剂 玻璃清洗剂 P3-SILIRON LS 碱性粉末清洗剂 VR 6334-16 碱性粉末脱脂剂 RIDOLINE 1559 S5 碱性粉末脱脂剂 RIDOLINE 336 碱性粉末脱脂剂 RIDOLINE 53 碱性清洗剂 P3-T714 碱性清洗剂 P3-T7221 碱性通用清洗剂 P3-SAXIN 碱性脱脂剂 PARCO CLEANER 1523 R 碱性脱脂剂 RIDOLINE 1559 碱性液体清洗剂 NOVACLEAN 6000 玻璃清洗剂 卷材用清洗 钢铁磷化前清洗 铝材用,适用于喷淋 压铸铝材专用 钢铁磷化前清洗 铜材专用清洗 不锈钢专用清洗 钢铁磷化前清洗 钢铁磷化前清洗 电子产品清洗 25KG 25KG 25KG 25KG 25KG 25KG 25KG 25KG 30KG 25KG 25KG 35KG 25KG 镁合金专用 镁合金专用 镁合金专用 镁合金专用 25KG 35KG 25KG 35KG 铝材用,进口,美国军标指定产品 铝材用,黄色膜层 铝材用,绿色三价铬膜层,须与 407 合用 铝材用,绿色三价铬膜层,须与 47 合用 铝材用,黄色膜层,配槽专用 铝材用,无色膜层 卷材用,无色膜层,无须后道水洗 铝材用,不含铬,环保产品 铝材用,不含铬,环保产品,配槽专用 27.2KG 30KG 25KG 25KG 30KG 25KG 25K G 25KG 25KG 用途说明 包装规格
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
/Langmuir©2010American Chemical SocietyHierarchical Carbon Foams with Independently Tunable Mesopore andMacropore Size DistributionsAdam F.Gross*and Andrew P.NowakHRL Laboratories,LLC,3011Malibu Canyon Road,Malibu,California90265Received February23,2010.Revised Manuscript Received April28,2010Hierarchical carbon foams with independently tunable mesopore and macropore size distributions were formed in a high internal phase emulsion(HIPE)template.The HIPE consists of an internal oil phase that controls the macropore dimensions and an aqueous resorcinol-formaldehyde precursor solution external phase that directs the mesopore size distribution.Once the emulsion is formed,the precursor solution is cured,fluid elements are extracted from the monolith via solvent exchange, and then the sample is pyrolyzed to create a hierarchical open-cell foam consisting of macropores with mesoporous carbon xerogel walls.Both mesopore and macropore size distributions may be independently tuned by changing the synthesis parameters.These samples have a peak in the mesopore size distribution that may be tuned to between5and8nm and macropore average diameters that may be tuned to between0.7and2.1μm.Furthermore,the0.7and2.1μm average diameter macropores have0.18and0.53μm diameter macropore windows between adjacent pores,respectively.Pore volumes up to 5.26cm3/g and electrical conductivities as high as0.34S/cm are observed after1200°C carbonization of the framework.These foams may have potential applications as3-D current collectors in batteries and as fuel cell catalyst supports.IntroductionMany porous materials may be formed from emulsion-templated solutions.1,2Emulsions are an advantageous tool for structural control because of their ease of fabrication,wide range of possible pore sizes,and scalability.Furthermore,porous silica,metal oxides, carbons,and metals have been templated with emulsions and these materials demonstrate the flexibility of this technique to form solid materials.1We are particularly interested in forming porous mate-rials in high internal phase emulsions(HIPEs)that have greater than74%internal phase volumes,which is the free space occupied by packed uniform spheres.3,4At higher than74%internal phase volume,the droplets become abutted and the degree of interconnec-tion between resulting pores increases.By forming solid materials in the continuous phase of a HIPE,an open cell foam monolith consisting of polymers,silica,or carbons may be produced.1-3,5-7 Furthermore,when the solid material formed in the external phase of the HIPE is mesoporous silica,a hierarchical macroporous and mesoporous foam with increased surface area and pore volume is obtained.6,7Our goal is to produce hierarchical macroporous carbons with mesoporous carbons walls made from inexpensive and easily removable fluidic precursors templated in HIPEs where both the mesopore and macropore dimensions may be indepen-dently tailored by altering the synthetic composition.Porous carbon materials with hierarchical macropores and mesopores have potential applications as3-D current collectors,electrodes,and catalyst supports.1,2,8-10The carbon framework provides electrical conductivity,and the macropore dimensions and interconnectivity provide control of mass transport and pore volume.1Furthermore,the mesopore structure influences the sur-face area,which is important for catalyst supports,and enables additional ion-transport channels in batteries.Finally,the carbon structure is chemically inert and will survive a variety of reaction conditions.There are many templating approaches that produce hierarchical porous carbons with different degrees of pore structure tunability.A porous carbon may be formed by carbonizing a HIPE in which the continuous phase consists of a polymerized styrene monomer.5This approach produces porous carbons with nan-ometer length scale pores in the walls that are induced as a result of densification during carbonization;however,no peak in the mesopore size distribution was shown because this approach does not actively template mesopores during synthesis.Additionally, hierarchical foams may be formed from agglomerated colloids of a carbonaceous precursor.11,12The packed colloids form macropores whereas surfactant templating in the colloids,or their densification during carbonization,produces mesopores.Pluronic surfactants may also be mixed with water and phenolic resins to form a macroporous and mesoporous hierarchical carbon structured by phase separation during hydrothermal treatment.13In another approach used by many groups,a resin may be polymerized around solid micrometer-sized polymer or silica colloids and then carbonized to form a hierarchical carbon.1,2,8-10,14-18Macropores*Corresponding author.E-mail:afgross@.(1)Zhang,H;Cooper,A.I.Soft Matter2005,1,107.(2)Yuan,Z.;Su,B.J.Mater.Chem.2006,16,663.(3)Cameron,N.R.Polymer2005,46,1439.(4)Barbetta,A.;Cameron,N.R.;Cooper,mun.2000,221.(5)Wang,D.;Smith,N.L.;Budd,P.M.Polym.Int.2005,54,297.(6)Hu,Y.;Nareen,M.;Humphries,A.;Christian,P.J.Sol-Gel Sci.Technol. 2010,53,300.(7)Carn,F.;Colin,A.;Achard,M.F.;Deleuze,H.;Sellier,E.;Birot,M.;Bakov, R.J.Mater.Chem.2004,14,1370.(8)Yu,J.-S.;Kang,S;Yoon,S.B.;Chai,G.J.Am.Chem.Soc.2002,124,9382.(9)Chai,G.S.;Yoon,S.B.;Yu,J.S.;Choi,J.H.;Sung,Y.E.J.Phys.Chem.B 2004,108,7074.(10)Lee,K.T.;Lytle,J.C.;Ergang,N.S;Oh,S.M.;Stein,A.Adv.Funct. Mater.2005,15,547.(11)Carriazo,D.;Pico,F.;Gutierrez,M.C.;Rubio,F.;Rojo,J.M.;del Monte,F.J.Mater.Chem.2010,20,773.(12)Zou,C.;Wu,D.;Li,M.;Zeng,Q.;Xu,F.;Huang,Z.;Fu,R.J.Mater. Chem.2010,20,731.(13)Huang,Y.;Cai,H.;Feng,D.;Gu,D.;Deng,Y.;Tu,B.;Wang,H.;Webley, P.A.;Zhao,mun.2008,2641.(14)Zhao,J.;Cheng,F.;Yi,C.;Liang,J.;Tao,Z.;Chen,J.J.Mater.Chem. 2009,19,4108.(15)Wang,Z.;Kiesel,E.R.;Stein,A.J.Mater.Chem.2008,18,2194.(16)Lukens,W.W.;Stucky,G.D.Chem.Mater.2002,14,1665.(17)(a)Baumann,T. F.;Satcher,J.H.Chem.Mater.2003,15,3745.(b)Baumann,T.F.;Satcher,J.H.J.Non-Cryst.Solids2004,350,120.(18)Woo,S.W.;Dokko,K.;Sasajima,K.;Takei,T.;Kanamura,K.Chem. Commun.2006,4099.Gross and Nowak Articleform after removal of the colloids,and their dimensions are adjusted by changing the initial colloid diameter.Further-more,very uniform macropores were created by first forming a colloidal crystal(instead of using agglomerated colloids)and then using this ordered solid as a template for the carbona-ceous material.2,8-10,14,17-19An additional level of order in hierarchical carbons may be induced by arranging smaller colloids around larger colloids in a templating colloidal crystal.2,18,19Mesoporosity is induced without a template in many of these colloid-templated carbons as a result of carbo-nization and densification of the resin.In a few reports,the mesopore dimensions were more precisely controlled when larger colloids were surrounded by a mesoporous carbon instead of a resin.16,17One drawback with solid colloidal particle templating is that it requires the initial synthesis of particles and creating porosity in these materials may necessi-tate subsequent etching with hydrofluoric acid if silica-based materials are used.Finally,a macroporous polymer foam may be infiltrated with mesoporous silica in order to form a hierarchical carbon.20The macropore size is controlled by the initial polymer foam pore dimensions,and the mesopore dimensions are controlled by the synthesis parameters of the mesoporous silica.Mesoporous carbon is formed through the vapor deposition of a precursor on the silica,carbonization of the precursor,and the removal of the silica template with HF. Our goal is to fabricate hierarchical porous carbon monoliths with independently tunable mesopore and macropore dimensions without templating from solid materials.By using an oil-in-water soft matter template,we are able to form an open-cell hierarchical carbon foam using inexpensive and easily removable templating materials.We template our materials with a high internal phase emulsion formed by the high-speed mixing of a silicone oil internal phase and an aqueous continuous phase.The aqueous phase contains a resorcinol-formaldehyde precursor solution that enables the tuning of mesoporosity with synthetic variables.21,22 Additionally,the macropore dimensions are tuned with oil viscosity.23A carbon foam is fabricated by blending the oil and aqueous phases,thermally curing the precursor solution,remov-ing the oil phase via solvent extraction,exchanging water for organic solvent,drying the monolith,and finally carbonizing the sample.This process results in a hierarchical macroporous foam made from mesoporous carbon xerogel.Our material differs from carbonized polymer HIPEs because the meosporosity is con-trolled by the xerogel synthetic variables rather than being a byproduct of polymer densification during pyrolysis.We believe that our material is the first hierarchical carbon monolith formed from all fluidic precursors with tunable pore size mesoporous xerogel walls.In this study,the composition of the aqueous and oil phases of the HIPE are varied to demonstrate independent tuning of the mesopore and macropore pore size distributions followed by the characterization of the resulting foams.Finally, we measure the electrical conductivity of the carbon foam mono-liths to determine if these materials could be candidates for fuel cell and battery applications.Experimental SectionAll chemicals were purchased from Aldrich and used as received.Resorcinol-formaldehylde precursor solutions were synthesized by mixing a2:1:0.002molar ratio of formaldehyde (37wt%solution in water)to resorcinol(99%)to sodium carbonate(99%)in deionized water.22The resorcinol-formalde-hyde precursor solutions contained30or40wt%organic content (formaldehydeþresorcinol mass).The resorcinol-formaldehyde precursor solutions were stirred until they dissolved and allowed to rest for1h before use in carbon foam fabrication.Carbon foams were fabricated by dissolving0.360g of sodium dodecylbenzenesulfonate surfactant(technical grade)in12.5g of a resorcinol-formaldehyde precursor solution that was then com-bined with37.5g of silicone oil(100and1000cP viscosity,Fluka brand DC200).The mixture was transferred to a Waring laboratory blender and blended for10min in all experiments.A white viscoelastic emulsion was obtained and transferred in equal fractions to three60mL Nalgene polypropylene jars.The jars were sealed and aged for72h at85°C,which resulted in bright orange resorcinol-formaldehyde foam monoliths formed in the shape of the Nalgene containers.The foams were then soaked in chloroform to extract the silicone oil.The chloroform bath was poured off and refilled with fresh solvent twice,and there was at least a4h soak time during each of the first two soaking cycles and 8h during the third cycle.The foams then were then removed from chloroform and placed in an acetone bath to exchange solvent for water in the monolith.The acetone bath was poured off and refilled two times with at least2h between each soaking cycle. Finally,the foam monoliths were removed from the acetone bath, allowed to dry in air,and heated in a tube furnace under flowing nitrogen from room temperature to800°C at2.6°C/min and maintained at800°C for6h to pyrolyze the resorcinol-formalde-hyde gel.In one case,a foam was pyrolyzed under flowing nitrogen and heated from room temperature to1200°C at2°C/ min and maintained at1200°C for6h in order to measure the effect of different pyrolysis conditions.There are no significant chemical hazards encountered during the preparation of the carbon foams described in this study.Samples carbonized at800°C are denoted throughout this article on the basis of the organic loading of the xerogel and the viscosity of the silicone oil.For example,a40%/1000sample was made by blending a40wt%organic resorcinol-formaldehyde precursor solution with1000cP silicone oil.The water/oil volume ratios of the40%/100,40%/1000,and30%/1000foams were 23.5:76.5,23.5:76.5,and23.9:76.1,respectively.The ratio is slightly higher for the30%/1000emulsions as compared to that of the40%/100and40%/1000emulsions because of the lower density of the30wt%resorcinol-formaldehyde precursor solu-tion.If the sample was carbonized at a temperature other than 800°C,then the carbonization temperature is listed after the orga-nic loading of the xerogel and the viscosity of the silicone oil. The sample pore size,surface area,and pore volume were characterized with N2adsorption and Hg intrusion at Micro-meritics Analytical Services(Norcross,GA).N2data were ana-lyzed using the Brunner-Emmett-Teller(BET)and Barrett-Joyner-Halenda(BJH)methods.24Hg intrusion experiments were sensitive to pores between3nm and360μm in diameter. SEM images were acquired on a Hitachi S-4800.Electrical conductivity was measured with an HP34401A multimeter using a four-wire probe.Current and voltage probes were placed on opposite faces of a carbon foam monolith,and the resistance was measured.This resistance was converted to conductivity by inverting the resistance value and dividing by the thickness of the monolith.Results and DiscussionCarbon foams were synthesized as described in the Experi-mental Section from a HIPE template consisting of an external(19)Chai,G.S.;Shin,I.S.;Yu,J.S.Adv.Mater.2004,16,2057.(20) Alvarez,S.;Fuertes,A.B.Mater.Lett.2007,61,2378.(21)Al-Muhtaseb,S.A.;Ritter,J.A.Adv.Mater.2003,15,101.(22)Li,W.;Lu,A.;Weidenthaler,C.;Sch€u th,F.Chem.Mater.2004,16,5676.(23)Mason,T.G.;Wilking,J.N.;Meleson,K.;Chang,C.B.;Graves,S.M. J.Phys.:Condens.Matter2006,18,R635–R666.(24)Lowell,S.;Shields,J.E.;Thomas,M.A.;Thommes,M.Characterization of Porous Solids and Powders:Surface Area,Pore Size and Density;Springer: Dordrecht,The Netherlands,2006.Article Gross and Nowak phase resorcinol-formaldehyde precursor solution and an internaloil phase.Carbon xerogel is formed in the external phase,and themesoporosity is controlled by the resorcinol-formaldehyde pre-cursor solution composition.The water fraction of the resorcinol-formaldehyde precursor solution acts as a porogen and templatesthe mesopores.Additionally,the organic fraction and amountof catalyst in the resorcinol-formaldehyde precursor solutioncontrol the micropore structure and shrinkage during dryingand pyrolysis.(Smaller amounts of catalyst relative to resorcinolcreate microporosity in the carbon particles that make up thexerogel and better resist shrinkage during drying.21)Althoughthe cured resorcinol-formaldehyde gel from the continuousphase of the HIPE provides structural rigidity in the car-bon foam,the oil phase creates regions where the resorcinol-formaldehyde precursor solution is excluded to produce macro-pores.3Additionally,the oil droplets are abutted,which resultsin windows forming between the macropores and creates an open-cell foam.We refer to these openings between macropores as“macropore windows”to differentiate them from the largermacropores.A conceptual diagram of the HIPE is shown inFigure S1.The HIPE that templates the carbon foam is formed using highshear mixing,and both the macropore and macropore windowdimensions are controlled by the oil viscosity.Higher oil viscosityresults in increased viscous resistance against oil droplet deforma-tion and separation into larger droplets.Thus a lower interfacialsurface area is created between oil and water for a given amountof mechanical energy deposited into the system(with the samemixing speed used in all experiments).25This reduced surface arearesults in larger droplets,larger macropores,and larger macro-pore windows.We varied both the oil viscosity and resorcinol-formaldehyde precursor solution composition to demonstratecontrol of both mesopore and macropore textural properties.Sample Fabrication.The monolithic structures of an as-synthesized resorcinol-formaldehyde HIPE-templated foam andof a subsequently800°C pyrolyzed carbon foam are shown inFigure S2.The foams take the shape of the cylindrical jars inwhich the emulsions are cured and maintain this shape with someshrinkage during carbonization.SEM images of the carbon foamsare shown in Figures1(top)and2and demonstrate that the foamretained the HIPE structure through carbonization.The macro-pores reproduce the morphology of the templating silicone oilphase with macropore windows resulting from abutted droplets.Similar openings between macropores are also observed incolloidally templated carbons because adjacent templates aretouching.2,8-14,17,18The higher-magnification image of themacropore wall in Figure1(bottom)clearly shows the hierarchi-cal structure of the mesoporous carbon xerogel forming the wallsof the macropores.SEM images in Figures2and S3of the30%/1000and40%/1000materials show larger macropores than observed in the40%/100sample and demonstrate that the macropore size may be tuned with the silicone oil viscosity. Higher-resolution images of the xerogel structure,such as in Figure1(bottom),show no discernible differences for all three samples.Additionally,multiple SEM images for each sample were used to calculate the mean macropore diameter by averaging the diameters of at least60macropores.When100cP silicone oil is used in the40%/100foam,the macropores have a mean diameter of0.7μm,whereas both the30%/1000and40%/1000samples have a mean macropore diameter of2.1μm.This represents a 3-fold increase in the macropore mean diameter from a10-fold increase in oil viscosity.To determine if the macropore pore diameters between samples were distinct,the statistical signifi-cance between the means of macropore distributions was mea-sured by applying Welch’s t test for unequal variances for all sample pairs.Differences in the means for each macropore distribution with differing oil viscosity(40%/100vs30%/1000 and40%/100vs40%/1000)were both found to be statistically different(two-tailed p<0.0001)whereas macropore populations between identical oil viscosity samples(30%/1000vs40%/1000) were not(two-tailed p=0.97).Porosimetry.The carbon foams fabricated in this study were characterized with Hg intrusion and N2adsorption to understand the pore structures.Hg intrusion is most sensitive to pores with diameters>50nm whereas the small size and gaseous delivery of N2atoms make their adsorption best suited to characterizing mesopores and micropores.24Pore size distributions for all800°C Figure1.SEM images of a40%/100carbon foam showing the HIPE-templated structure of the carbon foam.Scale bars are shown in the images.Macropores and macropore windows formed from abutted silicone oil droplets are observed in the top image. This image is indistinguishable from a HIPE-templated polymer, demonstrating that the original emulsion structure is retained.3 The mesoporous xerogel structure of the walls is observed under higher magnification(bottom).Figure2.SEM images of40%/1000(top)and30%/1000(bottom) carbon foams.Scale bars are shown in the images.The structure of a high internal phase emulsion is observed in both samples.(25)Meleson,K.;Graves,S.;Mason,T.G.Soft Mater.2004,2,109.Gross and Nowak Articlepyrolyzed sample were derived from Hg intrusion data(Figure S4)and are shown in Figure3.The peak positions for the30%/ 1000and40%/1000samples are both at0.53μm,and the peak for the40%/100sample is at0.18μm.Additionally,the pore size distributions for30%/1000and40%/1000samples overlap almost perfectly and are insensitive to resorcinol-formaldehyde precursor solution composition.We note that these pore dimen-sions are much smaller than those observed in SEM,which is due to Hg intrusion measuring the smallest openings between pores.24 These openings are the macropore windows seen in Figures1,2, and S3,and the macropore window dimensions observed in SEM images are in good agreement with the Hg intrusion data.In addition,the macropore window diameters have the same∼3-fold increase in diameter from a10-fold increase in oil viscosity observed for macropores in SEM.Because macropore and macropore window dimensions were tuned with silicone oil viscosity but not with the resorcinol-formaldehyde precursor solution,we conclude that the silicone oil viscosity alone mainly controls macropore and macropore window structure.All textural parameters from Hg intrusion are presented in Table1.All carbon foams have large pore volumes and porosities; however,the40%/100sample is the most distinct from the other samples.The40%/100sample is templated with a volume of silicone oil that is equal to that used for other samples;however,it is formed from lower-viscosity oil that is sheared into smaller droplets with an increased surface area to volume ratio during HIPE formation.The greater surface area of the smaller oil droplets should produce a carbon foam with greater surface area, which agrees with the data in Table1.Additionally,the smaller droplets result in the aqueous phase being distributed over a larger interfacial area,which yields thinner xerogel walls.The larger Hg intrusion pore volume of the40%/100sample may result from abutted droplets more easily producing macropore windows between thinner walls and creating more open space as compared to the thicker walls in the40%/1000and30%/1000samples.N2adsorption BJH pore size distributions calculated from N2 adsorption isotherms(Figure S5)are shown in Figure S6for all 800°C pyrolyzed samples.The40%/100and40%/1000samples have very similar pore size distribution peak positions,and larger pores are found in the30%/1000sample as shown by the1.33-fold larger pore size distribution peak position.This demonstrates that the composition of the aqueous phase,and not the templating oil, mainly controls mesopore pore size distribution peak positions. We note that the oil viscosity has some effect on mesopore structure as shown by comparing the100and1000cP oil-templated samples in Figure S6.The40%/100sample has some mesopore volume above20nm,but the pore volumes of the40%/ 1000and30%/1000samples are negligible in this region.This may be explained by the thinner macropore walls in the40%/100 sample having less internal volume as compared to that in the 40%/1000and30%/1000samples.There will be more xerogel material exposed to the oil-water interface in the40%/100 sample,and with fewer surrounding carbon particles on the interface,some surface pores may have diameters greater than 20nm.The calculated N2adsorption textural parameters of all samples are presented in Table2.All foams are microporous as well as mesoporous.The presence of micropores is confirmed by the t-plot micropore volume derived from the N2adsorption data(Table2).This micropore volume is formed during pyrolysis in the carbon particles that make up the mesopore walls.26Thus,these carbon foams have two levels of hierarchy:(1)the microporous carbon particles that make up the mesoporous xerogel and(2)the mesoporous xerogel composing the walls of the macropores. The data in Tables1and2demonstrates that the resorcinol-formaldehyde precursor solution controls the mesopore diameter but has little effect on the macropore and macropore window structure.Although the silicone oil viscosity controls the macro-pore volume and size,it minimally affects the peak position of the mesopore pore size distribution.Thus,the mesopore and macro-pore/macropore window dimensions in these hierarchical carbon foams may be independently adjusted.Furthermore,the macro-pore and macropore window average diameters should be tunable between0.7and2.1μm and0.18and0.53μm,respectively,by incorporating oil into the HIPE with the appropriate viscosity between100and1000cP(with200,350,and500cP currently available).Larger or smaller macropores and macropore win-dows than those made in this study are likely accessible using silicone oils with viscosity greater than1000cP or less than100cP. The mesopore size should also be tunable over a wider range of values than those shown in this study by using alternative aerogel or xerogel materials as discussed later in this article.We note that the mesopore pore size distribution peak posi-tions,pore volumes,and surface areas are significantly smaller than those observed in neat xerogels made with the same recipe. (These materials have also been referred to as aerogels in the literature,and they have sample compositions and processing conditions that are similar to those of our materials.22,27)The mesopore textural parameters may have been altered by the curing temperature profile or the presence of surfactant.The carbon foams in this study were cured for3days at85°C in contrast to the1day at room temperature,1day at50°C,and1 day at90°C previously used for a neat xerogel in ref22.When a neat40wt%organic xerogel control solution was cured for3 days at85°C and solvent exchanged with chloroform and acetone in an identical manner to that for the carbon foams,we obtained the same pore volume of1.39cm3/g and a similar surface area of 641m2/g as found in a traditionally cured sample.28However,the peak position in the pore size distribution increased from25 to51nm.Thus,the different curing temperature profile does alterFigure3.Hg intrusion pore size distributions of800°C pyrolyzedcarbon foams.Samples are indicated by the legend on the graph.Hg intrusion is sensitive to the smallest opening between pores,andthis data is indicative of the macropore windows.The40%/1000and30%/1000carbon foams were templated with higher-viscositysilicone oil than was the40%/100sample,and this results in largermacropores and macropore windows.This data demonstrates thatthe macropore window pore size is insensitive to the composition ofthe resorcinol-formaldehyde precursor solution and is tunable withsilicone oil viscosity.(26)Gavalda,S.;Gubbins,K.E.;Hanzawa,Y.;Kaneko,K.;Thomson,K.T.Langmuir2002,18,2141.(27)Wu,D.;Fu,R.;Dresselhaus,M.S.;Dresselhaus,G.Carbon2006,44,675.(28)Gross,A.F.;Vajo,J.J.;Van Atta,S.L.;Olson,G.L.J.Phys.Chem.C2008,112,5651.Article Gross and Nowakthe pore structure and probably contributes to differences in morphology,but it cannot explain the decrease in the pore volume,surface area,or pore size distribution peak position found in Table2.Another possible reason for the differences in mesopore morphology is that our samples contain2.8wt% sodium dodecylbenzenesulfonate surfactant in the resorcinol-formaldehyde precursor solution,which is not present in neat carbon xerogel syntheses.Equal and lower surfactant levels are known to densify aerogels/xerogels and result in lower pore volumes,pore diameters,and surface areas than observed in unmodified materials.27,29Thus,the large amount of surfactant in the HIPEs is likely the primary cause of the morphological changes that we observe in the xerogel framework.In the HIPEs, a significant fraction of surfactant is isolated at the oil-water interface and is not available to alter xerogel structure.Because of uncertainty about the distribution of surfactant between the oil/ water interface and the aqueous phase,we chose not to run a control experiment using only surfactant and our resorcinol-formaldehyde precursor solution because the interpretation of results would be not be meaningful.In addition to the previously discussed samples,we attempted to form a30%/100material for this study and obtained a resorcinol-formaldehyde foam that partially collapsed after sol-vent exchange and ambient drying.We measured a Hg intrusion pore volume of only1.30cm3/g in the30%/100carbonized foam, and the macropore walls buckled during drying as shown in the SEM image in Figure S7.Because lower organic loading xerogels contain less material to resist shrinkage and because the macro-pore walls are thinner in samples made with100cP silicon oil,it is understandable that the30%/100sample collapsed.This indi-cates that our method of forming carbon foams is limited by the strength of the xerogel walls.We believe our method could overcome these limitations and produce macroporous carbon foams with a larger range of densities and average mesopore diameters by incorporating supercritically dried carbon aerogel formulations or by reducing the amount of surfactant during HIPE fabrication.27,30Supercritically dried carbon aerogel for-mulations may allow the formation of lower-density carbon foams because of their lower organic content and gentler drying procedure.Additionally,reducing the surfactant concentration may allow larger mesopores.According to ref27,less surfactant in our xerogel formulation should increase the peak position of the mesopore size distribution up to values as large as25nm.This approach will work only if the surfactant preferentially migrates to the oil-water interface and sufficient surfactant is present to enable HIPE formation.A40%/100sample was pyrolyzed at1200°C where partial graphitization and sintering of the framework occur to determine the maximum possible electrical conductivity of our materials.21 After1200°C pyrolysis,the sample monoliths remained intact and SEM images in Figure S8shows that the macropore, macropore window,and xerogel structure were all maintained. Elevated firing temperatures are known to degrade the surface area and pore volume of xerogels and we characterized the porosity of this foam to quantify any reduced porosity.31-33 The macropore textural characteristics are shown in Table1, and a comparison of the macropore pore size distributions from 40%/100foams pyrolyzed at800and1200°C is shown in Figure S9.The40%/1001200°C sample shows a reduced macropore window diameter,which is expected because of the sintering of the carbon framework,and a slightly decreased foam density,which has been observed in another report on high-temperature-fired porous carbons.34However,the sample showed an unexpected large increase in macropore volume and surface area.We hy-pothesize that closed macropores originally formed around non-abutted oil droplets and may have opened as a result of the1200°C treatment of the carbon framework.The mesopore textural data for the40%/1001200°C sample is shown in Table2,and a comparison of the mesopore pore size distributions from40%/100foams pyrolyzed at800and1200°C is shown in Figure S10.The increase in the peak position of the mesopore size distribution and the reduced micropore volume fraction are both expected from the sintering of carbon particles that template the mesopores.21,31-33However,the vastly increased surface area and pore volume do not agree with known trends for surfactant-free carbon aerogels and xerogels.31,33The40%/100 1200°C sample is different from materials in previous reports on sintering effects because it contains surfactant in the synthesis solution,which results in denser networks of carbon particles forming the mesoporous framework.27,29The more closely packed carbon particles may form inaccessible pores,and the modifica-tions in the carbon framework that occur at1200°C may enableTable1.Calculated Textural Parameters of Carbon Foams from Hg Intrusion DatasampleHg intrusionsurface area(m2/g)Hg intrusionpore volume(cm3/g)foam density(g/cm3)skeletaldensity(g/cm3)porosity(%)Hg intrusion pore sizedistribution peak position(μm)40%/100155 4.370.197 1.41860.18 40%/1000102 3.270.263 1.88860.53 30%/1000118 3.220.262 1.68840.53 40%/1001200°C254 5.260.166 1.34870.15 Table2.Calculated Textural Parameters of Carbon Foams from N2Adsorption DatasampleN2adsorptionsurface area(m2/g)N2adsorption single-pointadsorption total porevolume(cm3/g)N2adsorption t-plotmicropore volume(cm3/g)N2adsorptionV micropores/V totalN2adsorption BJH poresize distribution peakposition(nm)40%/1001870.1930.0330.17 5.3 40%/10001740.1440.0350.24 5.7 30%/10002470.2490.0420.177.6 40%/1001200°C5930.5770.0680.127.6(29)Worsley,M.A.;Satcher,J.H.;Baumann,T.F.J.Non-Cryst.Solids2010, 356,172.(30)Liu,N.;Zhang,S.;Fu,R.;Dresselhaus,M.S.;Dresselhaus,G.J.Appl. Polym.Sci.2007,104,2849.(31)Zanto,E.J.;Al-Muhtaseb,S.A.;Ritter,J.A.Ind.Eng.Chem.Res.2002,41, 3151.(32)Gross,J.;Alviso,C.T.;Pekala,R.W.Mater.Res.Soc.Symp.Proc.1996,431, 123(also available at /bridge/product.biblio.jsp?osti_id=231318).(33)Lin,C.;Ritter,J.A.Carbon2000,38,849.(34)Wiener,M.;Reichenauer,G.;Hemberger,F.;Ebert,H.-P.Int.J.Thermo-phys.2006,27,1826.。