1q24.3~q25.3染色体片段缺失1例报告并文献复习
染色体1p32-p31缺失综合征1例报告并文献复习

染色体1p32-p31缺失综合征1例报告并文献复习万瑞平;朱晓丹;张美波;吴燕玲;黄小霏【期刊名称】《临床儿科杂志》【年(卷),期】2018(036)012【摘要】目的探讨染色体1p32-p31缺失综合征的的临床特征.方法回顾分析1例染色体1p32-p31缺失综合征患儿的临床资料,并复习相关文献.结果患儿,男,6个月,出生胎龄39+2周,出生体质量2.2 kg,出生时有轻度窒息,生后发育落后.患儿面容特殊,前额及后枕突出、眼距宽、眼裂小、双侧耳位低、低鼻梁、小鼻、唇薄、下颌小、高腭弓,右侧睾丸未降至阴囊,左侧阴囊积液,手指、足趾短小,四肢肌张力稍低.Gesell发育量表评估发育商46.头颅MRI示脑白质比例偏少,双侧侧脑室增宽,胼胝体后部纤细.脑电图示清醒背景较同龄儿略慢.脑干听觉诱发电位示左侧反应阈65 dB,右侧反应阈40 dB.高通量DNA测序发现患儿1p32.3-p31.3区域(chr1:54560001-66400000,hg19)存在缺失,缺失片段大小为11.84 Mb,包含NFIA基因在内的多个基因.确诊为染色体1p32-p31缺失综合征.结论染色体1p32-p31缺失综合征的临床特征包括胼胝体发育不良、脑室扩大/脑积水、巨头、特殊面容、不同程度发育迟缓,部分伴泌尿系统异常.NFIA基因是导致该病的关键基因.【总页数】4页(P908-911)【作者】万瑞平;朱晓丹;张美波;吴燕玲;黄小霏【作者单位】南方医科大学附属佛山妇幼保健院儿童康复科广东佛山 528000;南方医科大学附属佛山妇幼保健院胎儿医学研究所细胞遗传组广东佛山 528000;南方医科大学附属佛山妇幼保健院儿童康复科广东佛山 528000;南方医科大学附属佛山妇幼保健院儿童康复科广东佛山 528000;南方医科大学附属佛山妇幼保健院儿童康复科广东佛山 528000【正文语种】中文【相关文献】1.1q24.3~q25.3染色体片段缺失1例报告并文献复习 [J], 王依柔;李群;李辛;程青;李娟;王剑;沈亦平;王秀敏;沈永年2.X染色体连续缺失致慢性肉芽肿病和Mcleod综合征2例报告并文献复习 [J], 贺建新;郭雅洁;冯雪莉;王雷;徐保平;刘秀云;申昆玲;江载芳3.经寡核苷酸微阵列比较基因组杂交技术证实的12号染色体短臂中间缺失的病例报告及文献复习 [J], 卢洪涌;李晓军;黄宇烽;崔英霞;经卉;夏欣一;史轶超;杨滨;姚兵;戈一峰;梁泉4.Y染色体AZFb区sY1192缺失5例报告并文献复习 [J], 郭廷超; 孟令波; 梁悦; 卢永平5.染色体6p25缺失综合征1例报告并文献复习 [J], 潘翔;逯军;厉广栩;陈振因版权原因,仅展示原文概要,查看原文内容请购买。
一例染色体微缺失和微重复患儿的细胞遗传学及分子生物学分析

一例染色体微缺失和微重复患儿的细胞遗传学及分子生物学分析邱惠国;胡斌;洪国粦【摘要】目的确定1例畸形儿的核型,探讨二代测序(NGS)技术在分子细胞遗传学中的应用.方法对1例畸形儿、其父母及祖父三代人行外周血染色体G显带核型分析、NGS检测.结果 G显带染色体分析显示患儿、其父母及祖父核型均未见异常;NGS结果显示患儿46,dup(X)(q27.2q28):dup(Y)(p11.2p11.31);del(Y)(q11.223q11.23);del(9)(p23p 24.3),其父母及祖父结果均未见异常.结论通过NGS确定染色体微缺失及微重复是导致患儿多发畸形的主要原因.【期刊名称】《现代检验医学杂志》【年(卷),期】2019(034)003【总页数】3页(P34-36)【关键词】新一代测序(NGS);微缺失;微重复【作者】邱惠国;胡斌;洪国粦【作者单位】厦门大学附属第一医院检验科福建厦门 361000;厦门大学附属第一医院检验科福建厦门 361000;厦门大学附属第一医院检验科福建厦门 361000【正文语种】中文【中图分类】R446.7大片段(>10Mb)的染色体异常通常采用显带技术(如G显带)检测,而小片段(<10Mb)的染色体异常则不易通过显带技术检出[1]。
因此,常规的外周血染色体核型分析往往会漏检小片断平衡易位的携带者,或小片断的不平衡改变(微重复、微缺失)。
该研究应用新一代测序(next generation sequencing,NGS)技术对1例多发性畸形儿进行检测,以探讨NGS在核型诊断中的应用价值。
1 材料与方法1.1 研究对象患儿,男,出生4 h,以“生后吐沫,哭声低”为主诉入院。
患儿系第1胎,足月顺产。
患儿父母健康,非近亲婚配,否认家族遗传病病史。
体格检查:下颌畸形,哭声弱。
心脏彩超:右房右室增大,室间隔扁平(并发肺高压),三尖瓣中度返流,动脉导管未闭(2.0 mm),卵圆孔未闭,左室整体收缩功能早产低限。
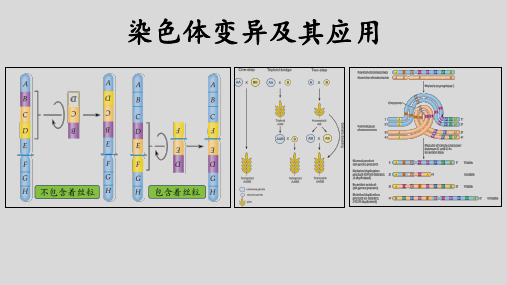
一轮复习染色体变异与育种PPT课件

染色体变异的控制措施
遗传筛查
对高风险人群进行遗传筛 查,及早发现染色体异常 ,采取干预措施。
遗传治疗
针对染色体异常引起的遗 传性疾病,采取基因治疗 、干细胞治疗等方法进行 治疗。
制定优生政策
通过制定优生政策,鼓励 优生优育,减少遗传性疾 病的发生。
05 复习与巩固
复习题
染色体变异
染色体变异的概念、类型和特点
巩固练习
选择题1
关于染色体变异的描述,正确的 是?
选择题2
在育种过程中,以下哪种方法可 以用于提高作物的抗病性?
巩固练习
• 选择题3:染色体变异对生物体的影响不包括?
巩固练习
填空题1
染色体变异可以分为哪几种类型?
填空题2
现代育种方法主要包括哪些?
巩固练习
简答题1
简述染色体变异对生物体的影响。
通过染色体变异,培育出 了转基因作物,提高了抗 虫、抗病能力。
03 染色体变异对生 物体的影响
染色体变异对生物体性状的影响
染色体变异可以导致 基因表达的改变,从 而影响生物体的性状 。
染色体变异可能导致 遗传性疾病的发生, 影响健康。
染色体变异可能导致 生殖细胞的异常,影 响繁殖能力。
染色体变异对生物体生存的影响
在育种方法方面,可以增加更多的实 际案例和应用,以增强学习的实用性 和趣味性。
THANKS
感谢观看
染色体变异可能导致生殖隔离 的形成,促进新物种的产生。
04 染色体变异的检 测与预防
染色体变异的检测方法
1 2 3
染色体数目变异检测
通过显微镜观察染色体数量是否正常,判断是否 存在染色体数目变异。
染色体结构变异检测
2025届高考生物一轮复习:染色体变异及其应用
方法一:进行无性生殖,将三倍体植株进 行组织培养获取大量培苗再移栽; 方法二:利用生长素或生长素类似物处理 二倍体植株未受粉的雌蕊,以促进子房发 育成无种子的果实,同时在花期全时段要 进行套袋处理,以避免受粉。
实例1:普通小麦的形成过程
这里的A、B、D代表的是染色体组,而不是基因 一粒小麦和山羊草是同一物种吗? AB是几倍体?ABD是几倍体?普通小麦是几倍体?
②三倍体西瓜无子的原因 三倍体西瓜在减数分裂过程中,由于染色
体联会紊乱,不能产生正常配子。 ③秋水仙素溶液处理的位置和原因?整个植株染色体都加倍了吗? 处理芽尖,因为芽尖是有丝分裂旺盛,用秋水仙 素处理可以抑制细胞有丝分裂时形成纺锤体,导 致细胞内染色体数目加倍,从而得到四倍体植株。
④据图分析你能说出产生多倍体的基本途径吗? 用秋水仙素处理萌发的种子或幼苗。
F1
杂交 高杆抗病DdTt
思考2:用秋水仙素处理单倍体一定 能够获得纯合子吗?
不花一药定离,体如培果养单≠单倍倍体体体育细种胞:中单基倍因体型育为种AB,
配子
DT Dt dT dt 经一秋般水包仙括素杂处交理、后花基药因离型体为培A养A、B秋B,水是仙纯素合
花药离体培养
子处;理如和果筛单选倍4个体过体程细,胞不中能基简因单型地为认A为a,花经药 秋离水体仙培素养处就理是后单基倍因体型育为种A的A全a部a,。是杂合子。
正常眼
棒眼
正常眼
棒状眼
3.倒位:
染色体的某一片段位置颠倒引起变异。 结果 :基因排列顺序发生改变
实例:果蝇卷翅的形成 染色体正常 染色体片段颠倒
正常翅
卷翅
倒位后的联会现象:
正常翅
散发性结直肠癌染色体3q24-25区杂合性缺失的精细定位

癌基 因。方法 将 3号染 色体采用 1 3个微卫星 D A标记 , 3 2 - N 在 q42 5区另取 6 个微卫星标记对 8 3例结直肠癌患
者 的肿瘤和正常组织进行 P R反应。P R产物在 A I rm 37自动荧光测序仪进行 电泳 3h 以 G nsa3 1和 C C B i 7 Ps , eecn . G nt e . eo pr 1软件进行基因分 型。结果 3号染色体 上发现 了 2个 高频杂合 缺失 区即 3 1 y 2 p 4区和 3 2 —5区 , q42 在 32- q 42 5区界定 了 1 个大约 2e m的跨越 D S 29位点的杂合缺失区。结论 3 17 3 2 -5区存在 1个精细 的杂合缺失 q42
so i n,s ek te ne t e h w umo s p e s r g ne i c lr ca u rg n ss M eho ห้องสมุดไป่ตู้Thite l mo p i mir s tli r u prs o e s n o oe tlt mo e e i . i t ds re n poy r he c o aelt e
ma k F e e u e o a ay e te e mo o ,a d a oh r r e sw r p l d o h o s me3 2 - 5 r e 8w r s d t n z  ̄o s me 3 l h n n t e ma k r ee a pi n c r mo o q 4 2 .Ca c ra d 6 e n e n
2 a h tr z g ss d p ein rgo c o s D3S 2 9 st s fn d o 2 25 Co ls o s Th r s a l s f h t m ee o y o i e lto e in a r s 1 7 i Wa de e n 3q 4- . us i ncu i n ee i o s o e— e o y o iy o h o s me 3 2 2 r z g st n c r mo o q 4- 5,whih mu tc n an o e o r mk o u r s p r s o e e n e l re a a e c s o ti n rmo e t n wn tmo u p s rg n s o oo e tlc e n -
人类的性别决定

人类的性别决定【篇一:人类性别决定】人类性别决定08生本(1)班 2008574117 梁柳清摘要:人类的性别是一个特别明显的性状,两性性状的差异不仅表现在外部形态特征上,也存在内部器官的结构上。
性别决定是一个复杂的生理现象,存在着一定的决定机制,这种决定机制主要来自于遗传物质(性染色体和基因)、激素、环境等因素。
人类性别决定相关基因的研究对性分化与发育异常等临床疾病的诊断和治疗具有十分重要的意义。
关键词:人类性别决定、基因按照最初级的认识,人类性别是由染色体决定的,即细胞染色体核型为46,xx的表现为女性,核型为46,xy的个体表现为男性。
但后来发现,有些人的细胞染色体核型相同(如都是46,xx或46,xy)而表现出来的性别却不同,即核型为46,xx的个体既有女性也有男性,同样核型为46,xy的个体既有男性也有女性。
为什么会出现这种奇怪的现象呢?对以上的现象有史以来存在着各种各样的猜测,但都没有科学的根据。
直到20世纪初,科学家发现了人体细胞内的染色体后,才揭开了性别的奥秘,人们对性别决定才有了新的认识。
性别决定和分化是个体正常生存和发育的必要一环。
染色体尤其是性染色体的完整性是性别决定的基础,并协同其他基因和激素共同指导着性器官的发育和成熟,最终形成睾丸或卵巢。
性别决定包括两方面内容:第一为性腺的决定和发育,称为初级性别决定;第二为附属性器官、特征及性行为的建立,称为次级性别决定。
前者受到多种基因的调节,占有优先和主导地位,后者的建立主要来自性腺产生激素的诱导和调节作用。
一、性别决定的调控性别决定的调控主要包括两个方面,即转录因子编码基因和常染色体基因调控。
1、性染色体基因调控:sry基因sry(sex-determiningregion y)在人类性别决定中起着关键作用,能启动睾丸分化,对睾丸发育负调节起抑制作用。
大量临床案例显示xy男性性逆转的患者其性染色体的sry基因发生了突变。
sry基因定位于yp11.23,是启动精巢发育的关键基因,控制胚胎向雄性方向发育。
产前诊断11q23.3-q25缺失1例

图1 胎儿 S NP — a r r a y检 测 结 果 , 1 1 号染色体示意 图( 红色表示缺失)
约为 l / 1 0 0 0 0 0 , 男 女 比例 约 为 1 : 2 l _ 1 ] 。i 9 7 3年 ,
丹 麦 的遗 传学 家 P e t r e a J a c o b s e n首次 对 1 1号 长 臂
【 中图分类号】 R 7 1 4 . 5 5
【 文献标识码】 B
1 病 例 资 料
HC 2 1 7 mm , A C1 9 8 mm , F L 4 3 mm。 胎 儿 室 间 隔 缺
损; 左 肾大 小 约 3 4 mm×1 8 mm, 肾 盂分 离 , 前 后 径 约 7 . 4 mm, 肾盏 扩 张 , 右 肾大 小 约 2 9 am× 1 r 4 mm, 肾盂 分离 , 前后 径 约 6 . 0 mm, 未 见 肾 盂扩 张 ; 肠 管 回声 增
末 端 缺 失 进行 了描 述 , 到 目前 为 止 , 全 球 已报 道 的
病例有 2 0 0多 例 。J a c o b s e n综 合 征 患 者 l 临床 表 现
图 2 羊 水 染 色体 核 型分 析 , 箭 头所 指 为 l 1 号 染 色体 缺 失
与缺 失 片 段 大小 与 密 切 相关 , 包括发育迟缓 , 精 神
患者女, 3 5岁 , G7 P 1 , 2 0 0 4年 剖 宫 产 1女 , 健 康, 既往 史无 特 殊 。末次 月经 : 2 O 1 5年 1 1月 i 0日。
孕2 4周 , 因 B超 发 现 胎 儿 异 常 到本 中 心 就 诊 。唐 氏筛 查 高风 险 , l 8 一 三体 风险值 1 : 1 0 7 。停 经 2 3 周 外
2q31.1微缺失综合征1例报告并文献复习

doi:10.3969/j.issn.1000-3606.2020.06.0142q31.1微缺失综合征1例报告并文献复习陈 静 田茂强 李 娟 束晓梅遵义医科大学附属医院小儿内一科(贵州遵义 563003)摘要: 目的 提高对2q31.1微缺失综合征基因型及表型的认识。
方法 总结分析1例2q31.1微缺失综合征患儿的临床资料并复习相关文献。
结果 女性患儿,11月龄,自幼全面发育落后伴惊厥2次,特殊面容,肢端畸形,四肢肌张力减低,指、趾畸形;头颅MRI示胼胝体发育不良。
应用染色体芯片检测技术,采用比较基因组杂交技术(array-CGH)证实2q31.1-2q31.3区域存在7.279 Mb微缺失:arr 2q31.1q31.3(174570453-181849708)×1。
患儿确诊为2q31.1微缺失综合征。
文献报道2q31.1微缺失综合征中HOXD基因簇及其调控序列的单倍体剂量不足导致肢端畸形;LNPK功能缺失性变异导致惊厥发作合并胼胝体发育不良的神经发育性疾病,表现为精神运动发育迟滞、智力障碍、肌张力减低、惊厥发作和胼胝体发育不全。
该患儿神经系统受累表现与LNPK单基因变异的表型相似,推测患儿神经系统受累可能由LNPK单倍体剂量不足导致。
结论 对全面发育落后合并肢端畸形者需警惕2q31.1微缺失综合征。
关键词: 2号染色体; 2q31.1微缺失综合征; 肢端畸形。
Nervous system involvement in 2q31.1 microdeletion syndrome caused by mutation in LNPK: a case report and literature review CHEN Jing, TIAN Maoqiang, LI Juan, SHU Xiaomei (Department of Pediatric Medicine, Affiliated Hospital of Zunyi Medical University, Zunyi 563003, Guizhou, China)Abstract: Objective To improve the understanding of genotype and phenotype of 2q31.1 microdeletion syndrome.Method The clinical data of 2q31.1 microdeletion syndrome in a child were analyzed, and the relevant literature was reviewed. Results An 11-month-old girl had global developmental delay and convulsions for 2 times since birth. She presented with special faces, deformity of extremities, decreased muscle tension of limbs, deformity of toes and fingers.MRI of the brain showed dysplasia of the corpus callosum. The microdeletions of 7.279 M b in 2q31.1-2q31.3 region: arr 2q31.1q31.3 (174570453-181849708) ×1 were confirmed by chromosome chip detection and comparative genomic hybridization (array CGH). The child was diagnosed with 2q31.1 microdeletion syndrome finally. It has been reported that the haploinsufficiency of HOXO gene cluster and its regulatory sequence in 2q31.1 microdeletion syndrome leads to limb deformity. The loss-of-function mutations of LNPK result in neurodevelopmental disorders with convulsive seizures and corpus callosum hypoplasia, characterized by psychomotor retardation, mental retardation, hypotonia, convulsive seizures and corpus callosum hypoplasia. The neurological involvement of this child is similar to the phenotype of LNPK single gene mutation. Thus, it is speculated that the neurological involvement of the child may be caused by haploinsufficiency of LNPK. Conclusion It is necessary to be aware of 2q31.1 microdeletion syndrome in patients with global developmental delay and limb deformity.Key words: chromosome 2; 2q31.1 microdeletion syndrome; limb deformity2q31.1微缺失综合征(2q31.1 microdeletion syndrome)最早于2011年报道,以肢端畸形、神经系统受累及其他内脏器官畸形为临床特点[1]。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
doi:10.3969/j.issn.1000-3606.2018.07.0171q24.3~q25.3染色体片段缺失1例报告并文献复习王依柔 李 群 李 辛 程 青 李 娟 王 剑 沈亦平 王秀敏 沈永年上海交通大学医学院附属上海儿童医学中心(上海 200127)摘要: 目的 分析临床罕见的1号染色体片段缺失的临床特征以及基因特点。
方法 回顾1例1号染色体片段缺失伴重度矮小以及生长发育落后患儿的临床资料,并复习相关文献。
结果 患儿,男,3岁。
宫内发育迟滞,出生后匀称性矮小并有特殊面容,伴多发畸形(短指、宽指、小头畸形等)、隐睾、小阴茎、语言发育迟缓。
染色体核型分析为46,XY,染色体结构未见异常。
基因芯片检测显示1号染色体q24.3~q25.3区域存在一段大小为14 615 kb的杂合缺失。
结论 患儿致病原因为1号染色体q24.3~q25.3区域存在的大小为14 615 kb的杂合缺失。
关键词: 身材矮小; 发育落后; 多发畸形; 1号染色体片段缺失Chromosome fragment 1q24.3-q25.3 deletion:a case report and literature review WANG Yirou, LI Qun, LI Xin, CHENTG Qing, LI Juan, WANG Jian, SHEN Yiping, WANG Xiumin, SHEN Yongnian (Shanghai Jiao Tong University, School of Medicine, Shanghai Children’s Medical Center, Shanghai 200127, China)Abstract: Objective To investigate the clinical features and genetic characteristics of deletions of the long-arm of chromosome 1 which is rare seen clinically. Methods The clinical data from one child with chromosome 1 deletion who is severely dwarf and has development delay were analyzed, and relevant literatures were reviewed. Results Three-year-old boy had intrauterine growth retardation, postnatal growth restriction and special face, with multiple malformations (short finger, wide finger, small head deformity, etc.), cryptorchidism, small penis, language retardation. Chromosomal microarray analysis results demonstrated a 14615 kb heterozygous deletion in 1q24.3-1q25.3. Conclusions The 14615 kb heterozygous deletion in 1q24.3-1q25.3 is the pathogenic factor in this child.Key words: short statues; development delay; multiple malformations; del(1q)目前为止,对于1号染色体长臂缺失的报道仍极为罕见[1]。
1980年Schinzel等[2]根据缺失部位将1号染色体缺失分为3种类型:1q近端缺失(1q21~22→q25)、1q中间缺失(1q24~25→q32)以及1q远端缺失(1q42→末端)。
其中1q中间缺失型已报道40余例,但缺乏共同的特征性表型[3]。
近年来,Burkardt等[4]提出1q24q25微缺失综合征的概念,即伴有1q24q25缺失的患者,具有共同的特征性临床表现,包括出生前及出生后的生长发育迟缓、短指畸形以及特殊面容(上眼睑肥厚、异常小耳朵、短鼻伴球状鼻尖、帐篷样上唇以及小颌畸形等)。
随着基因诊断技术的发展,对染色体畸变的诊断更为准确,这类患者的报道也逐渐增多。
本研究回顾分析1例重度矮小伴生长发育落后、特殊面容伴多发畸形,经全基因组芯片检测提示1号染色体q24.3~q25.3存在14 615 kb杂合缺失,并伴1q24q25缺失患儿的临床特点及基因检测结果,并与既往报道的病例进行对比和分析。
1 临床资料患儿,男,汉族,3岁3个月,因身材矮小伴发育落后就诊。
患儿体质量9 kg,身高74 cm,均位于同年龄、同性别儿童P3以下;现刚会独立行走,有时仍需搀扶,尚不会说话(只会发无意义的单字)。
患儿系G2P2,孕40周出生,出生身长41 cm(男童出生身长-3SD 为45.2 cm),出生体质量1 760 g(男童出生体质量-3SD为2.26 kg[5]),为足月小样儿。
出生后母乳喂养,婴幼儿期生长发育落后,根据随访记录,将患儿生后5个月、9个月、1岁、2岁的身高、体质量,绘制生长曲线图显示,身高、体质量始终位于同年龄、同性别儿童生曲线图的P3线以下(图1)。
于2015年11月曾行左侧隐睾手术。
父母非近亲婚配,家族中无遗传代通信作者:王秀敏 电子信箱:wangxiumin 1019@谢类疾病史;有一姐姐,4岁7个月,生长发育正常。
入院体格检查:神清,精神反应可,浅表淋巴结无肿大;前发际偏高,眼距稍宽,鼻梁塌陷、宽鼻尖,下唇薄,下颌小,耳位低,短颈(图2A 、2B );双手手指粗短,皮肤稍黑,双侧掌纹呈“通贯手”;心肺腹无异常;腹股沟见隐睾术后疤痕,小阴茎;神经系统检查未见异常。
实验室检查中肝肾功能、甲状腺功能、性激素等均无异常;两次查类胰岛素样生长因子(IGF-1)分别为49 、54.1 ng/mL ,皮质醇7.5 ng/mL ,促肾上腺皮质激素21.10 pg/mL ;串联质谱检查所有氨基酸和酰基肉碱谱均无显著异常。
外周血染色体核型分析(550带,G 带)见46条染色体,数目正常,性染色体为XY ,染色体结构未见异常(图3)。
7月龄时GESELL 评分:运动能DQ 粗运动82 (相当于4个月)、细运动56(相当于3个月),应物能DQ 43(相当于2.3个月),言语能DQ 69(相当于3.7个月),应人能DQ 56(相当于3个月),均落后于正常。
头颅MRI 示轻度脑室周围白质软化,胼胝体偏薄,双侧额部脑沟增宽,双侧乳突积液。
阴囊B 超示双侧睾丸在位(术后);心脏彩超示卵圆孔未闭。
腹部B 超示肝脾、双肾、均偏小。
1岁11月龄骨龄平片示骨龄落后5~8个月(图2C )。
经父母知情同意,行细胞遗传学检查并完善全基因芯片扫描。
使用SurePrint G 3 Custom CGH+SNP (4*180K) 芯片进行全基因组扫描,发现患儿1号染色体q 24.3~q 25.3区域存在一段大小为14 615 kb 的杂合缺失(图4)。
A 、B. 特殊面容(前发际线偏高、弓形眉、眼距宽、宽鼻尖、鼻梁塌陷、小颌畸形等)、手指粗短、皮肤稍黑等;C. 2岁骨龄平片示手腕骨在15月龄左右,手指骨在18月龄左右图2 患儿特殊面容及骨龄平片表现图3 患儿外周血染色体核型分析结果见1号染色体q 24.3~q 25.3区域存在一段缺失图4 患儿全基因芯片扫描图2 讨论矮小症是儿科常见疾病,是指与同地区、同年龄、同性别正常儿童比较,身高低于正常儿童生长曲线的P 3[6]。
引起儿童矮小的病因繁多,与遗传、营养、环境、精神心理因素、宫内发育迟缓、下丘脑-垂体-胰岛素样生长因子1轴异常、染色体畸变、全身性慢性疾病、遗传代谢病以及内分泌激素等关系密切[7]。
在患儿出生早期寻找致生长迟缓病因是诊疗的 重点。
本例患儿出生时及出生后身高、体质量均低于同年龄、同性别儿童-3SD ,语言、行为以及智力发育均明显落后,还伴有隐睾、心血管系统畸形、通贯手、小颌畸形等。
考虑到染色体异常是引发先天畸形、精神疾患以及智力缺陷的常见原因[8],故对患儿外周血染色体核型进行分析,但结果为阴性。
再行全基因芯片检测,发现1号染色体q 24.3~q 25.3区域存在一段大小为14 615 kb (约14.3 Mb )的杂合缺失,该区域包含多个致病基因如ABL2、ACBD6、DNM3、CENPL、图1 患儿身高和体质量生长曲线图 身高 体质量ABCDARS2、LHX4、TNFSF4、PRDX6、XPR1等,其中LHX4基因位于1q25,在调控运动神经元的支配以及促进垂体发育过程中起作用[9]。
曾有报道1例家族性LHX4杂合突变,其表型特征为身材矮小,垂体前叶激素严重缺乏,垂体和脑后部发育缺陷以及蝶鞍畸形,缺失这一区域所导致LHX4单一不足可造成儿童生长激素的缺乏[10]。
IGF-1的生理作用是促进骨骼细胞的分化和增殖,介导生长激素对生长的促进作用。
本例患儿IGF-1始终处于同年龄层的低水平,提示可能存在生长激素缺乏,但其生长激素水平还需进一步的生长激素激发试验明确。
另一个可能导致该综合征的基因为DNM3[4],其是导致身材矮小、小头畸形以及神经发育障碍的主要基因之一[11]。
DNM3由18个外显子组成,编码含702个氨基酸的蛋白dynain-3[12]。
该蛋白在成人的大脑和脊髓中高表达,可与突触后Homer蛋白结合形成Shank-Homer复合物,起到促进谷氨酰胺能神经元成熟以及增加突触强度的作用[13]。
最新报道显示,在DNM3的内含子中有7 kb的反义转录本编码2种小分子RNA(miR199、miR214),在与面部颅骨发育相关的间质和软骨膜中高表达[14]。
研究发现,敲除miR199+214 的小鼠,虽然dynain-3蛋白的功能可以正常表达,小鼠仍表现为喂养困难、生长停滞、颅面发育不全以及骨质疏松等[15]。
本例患儿手掌X片示手小、短指和宽指,临床可见身材极度矮小、下颌小、眼距宽,在一定程度上反映了骨骼的改变。
此外,CENPL 基因位于1q25.1,编码着丝粒蛋白L,在着丝粒发挥正常功能和有丝分裂过程中起重要作用[16]。
CENPL 基因缺失可能是导致伴有1q24q25缺失患者生长发育迟缓和小头畸形发生的原因之一[4]。
通过回顾以往病例并进行归纳分析发现,在1号染色体长臂缺失中,伴1q24~q25缺失的患者拥有类似的表型。