双向电泳步骤--标准操作完整版
(完整版)双向电泳原理与流程

GE Healthcare Life Sciences ——实现从发现到功能研究,从体外到体内的突破
GE Healthcare milestones in 2-D electrophoresis
2002
Amersham Biosciences launched Ettan™ DIGE
1991
Pharmacia Biotech introduced Immobiline DryStrip
1973
1978
1982
LKB introduced Immobiline™
1979
Pharmacia Fine Chemicals introduced Pharmalyte™
The first multiple separation system, ISO-DALT, was developed
4/ GE Title or job number /
2/3/2020
Why 2DE?
Only “Proteomics” is the large-scale screening of the proteins of a cell, organism or biological fluid, a process which requires stringently controlled steps of sample preparation, 2-D electrophoresis, image detection and analysis, spot identification, and database searches.
2/3/2020
双向电泳详细操作过程
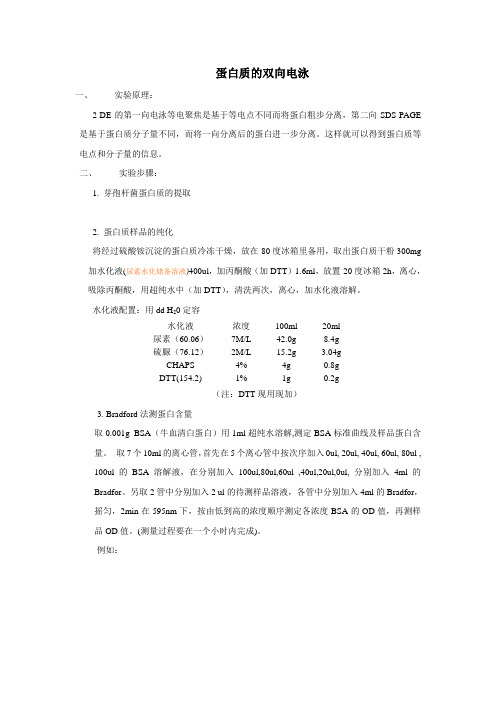
蛋白质的双向电泳一、实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE 是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二、实验步骤:1. 芽孢杆菌蛋白质的提取2. 蛋白质样品的纯化将经过硫酸铵沉淀的蛋白质冷冻干燥,放在-80度冰箱里备用,取出蛋白质干粉300mg 加水化液(尿素水化储备溶液)400ul,加丙酮酸(加DTT)1.6ml,放置-20度冰箱2h,离心,吸除丙酮酸,用超纯水中(加DTT),清洗两次,离心,加水化液溶解。
水化液配置:用dd H20定容水化液浓度100ml 20ml尿素(60.06)7M/L 42.0g 8.4g硫脲(76.12)2M/L 15.2g 3.04gCHAPS 4% 4g 0.8gDTT(154.2) 1% 1g 0.2g(注:DTT现用现加)3. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 20ul, 40ul, 60ul, 80ul , 100ul的BSA溶解液,在分别加入100ul,80ul,60ul ,40ul,20ul,0ul, 分别加入4ml的Bradfor。
另取2管中分别加入2 ul的待测样品溶液,各管中分别加入4ml的Bradfor,摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
例如:标准曲线方程式:Y= aX+b.其中Y为OD值,X为蛋白含量。
a、b通过作图输入数据可知G250的配置:称取G250 固体0.1g加水定容至1L。
使用前滤纸过滤。
比色皿用70%的乙醇保存,待用时用双蒸水冲洗,再用无水乙醇冲洗,双蒸水冲洗,再加入待测样品溶液润洗,然后,加入样品,测定OD值。
聚丙烯酰胺凝胶双向电泳

(8)平衡: 将推出的一条凝胶条放入盛有10ml平衡液的小烧杯中,
浸泡平衡20 min,然后进行第二向电泳. (9)染色:
另一条胶放入固定液中固定1~2 h,用考马斯亮蓝R250 染色1 h,再用脱色液脱色,观察第一向聚焦效果。
第二向电泳--SDS-聚丙烯酰胺凝胶电(SDS-PAGE) 1.溶液配制 (1) 30% Acrylamide第二向储液(500 mL/班) 含29.1% (w/v) acrylamide和0.9% (w/v) N,N'-methylene-bisacrylamide,配制方法同第一向储液。 (2) 10% SDS 称10 g SDS用重蒸水溶解,定容到100 mL, 室温存放。
3\操作步骤 ① 将胶置于150ml 40%甲醇-10%冰醋酸中浸泡,摇动 固定过夜。 ② 取出胶,置于120ml 10%甲醇-5%冰醋酸中浸泡,摇 动固定30min。 ③ 用去离子水将胶浸泡、漂洗,第一、二次,每次间隔 20min。 ④ 去离子水摇动漂洗,第三、四次,每次间隔30min换水 一次。 ⑤ 倾出去离子水,加入5μg/ml的DTT溶液120ml,摇动浸 泡30min。
2.实验步骤 (1)组装夹心式玻璃板、制胶支架底座调水平;
取一块长玻璃板和一块带磨口槽的短玻璃板,将短玻璃 板带磨口槽的一边位于上端与长玻璃相对,两玻璃板之间 的左、右两端各放入一条间隔条,对齐,插入玻璃板固定 夹,将上面的固定螺丝拧紧,将此夹心式玻璃板放入制胶 支架底座,插入凸轮,向内侧推入并旋转180º,夹心式玻 璃板即被固定。
聚丙烯酰胺凝胶双向电泳
(4)加样 待凝胶聚合后,用微量注射器吸去覆盖在胶上的 重蒸水。在每根胶柱上加20~30 µL蛋白质样品溶液,然后 再加入覆盖溶液至管口。
蛋白质双向电泳实验流程

蛋白质双向电泳实验流程一.样品制备1.研磨研磨时间要尽量短,并需及时补充液氮,研磨要充分,同时要保证损失少。
2.重新加入8mltris饱和状态酚(ph8.8)和8ml裂解液,在通风橱内研磨30s。
先加8mltris饱和状态酚,tris饱和状态酚会变为液态,此时需以研磨碓将液态的tris饱和状态酚研磨变成小块。
接着重新加入8ml裂解液,也须要将液态的裂解液研磨变成小块。
等三者搅匀后,将粉末迁移至45mltube。
3.振荡30min。
室温静置,等待tube中液态变为液体后,已经开始震荡。
震荡须要持续30min,每震荡1min,放在冰上加热1min。
4.10000g,4℃,10min。
将酚相(topphase)转移至45mltube。
酚相(topphase)可置于冰上。
酚二者必须就是绿色的,水相必须就是淡黄色的。
5.取6ml的抽提液和6ml饱和酚加入水相,蜗旋振荡30min。
振荡需持续30min,每振荡1min,置于冰上冷却1min。
6.10000g,4℃,10min。
将酚二者(topphase)迁移至45mltube。
7.沉淀酚相。
取一定体积(是酚相的5倍)的0.1m乙酸铵/甲醇溶液(c20℃保存)于酚相(45mltube)。
振荡30s,c20℃培育1h或过夜。
8.冲洗结晶①15min,20,000g,4℃。
弃上清。
②提10ml0.1m乙酸铵/甲醇溶液,用移液器吸打。
c20℃结晶30min。
③15min,20,000g,4℃。
弃上清。
④重新加入10ml乙酸铵/甲醇溶液,用移液器吸打。
c20℃结晶30min。
⑤15min,20,000g,4℃。
弃上清。
⑥提10ml80%丙酮(ice-cold),用移液器吸打。
c20℃结晶30min。
⑦15min,20,000g,4℃。
弃上清。
⑧重新加入10ml80%丙酮(ice-cold),用移液器吸打。
c20℃结晶30min。
⑨15min,20,000g,4℃。
弃上清。
双向凝胶电泳实验方法

双向凝胶电泳实验方法实验方案1.样品制备(Sample preparation)1.1仪器高速离心机1.2试剂NS(制备好的);Citrated human plasma ;溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base) 1.3样品处理方法1).吸取适量的制备好的NS加入到Citrated human plasma 中,在温度为37°下孵化5min。
NS与血浆的加入比例为0.6:8ml。
2)孵化后,将样品高速离心,离心转速为,时间为。
3)除去上清液,将沉淀的纳米球再混悬于0.05M,pH7.4的磷酸缓冲盐中,再次以上次条件离心,本步骤重复三次。
4)将离心后收集到的总蛋白溶解于溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base)。
2.第一相等电聚焦(IEF)2.1固定化 pH 剃度胶条的水化(IPG strip rehydration) 2.1.1仪器IPGphor2.1.2试剂水化液(8 M 尿素,2 % CHAPS,15 mM DTT 和 0.5 % IPG 缓冲液) 水化液需当天新鲜配制2.1.3实验步骤A. 加样品溶涨1).用样品溶解缓冲液(9M 尿素,4 , CHAPS,2 , IPG 缓冲液,40 mM DTT,40mM Tris-base)溶解样品。
蛋白质上样浓度(Q:如何确定上样浓度,)不要超过 10 mg/ml,否则会造成蛋白质的集聚或沉淀。
2).吸取适量(见下表1)含有样品的水化液放入标准型胶条槽中,为确保样品充分进入胶条中,不要加入过量的水化液。
表1 IPG胶条所需水化液体积3).去掉 IPG 胶条的保护膜,胶面朝下,先将 IPG 胶条尖端(阳性端)朝标准型胶条槽的尖端方向放入胶条槽中,慢慢下压胶条,并前后移动,避免生成气泡,最后放下 IPG 胶条平端(阴极),使水化液浸湿整个胶条。
双向电泳操作步骤

双向电泳操作步骤双向电泳操作步骤及相关溶液配置A(实验过程一实验原理:2-DE的第一向电泳等电聚焦是基于等电点不同而将蛋白粗步分离,第二向SDS-PAGE是基于蛋白质分子量不同,而将一向分离后的蛋白进一步分离。
这样就可以得到蛋白质等电点和分子量的信息。
二实验步骤:1. 样品的溶解取纯化后的晶体蛋白3.0mg,加入300ul裂解液(1mg蛋白:100ul裂解液)振荡器上振荡10min左右,共处理一个小时。
其中每隔10,15分钟振荡一次,然后13200rpm离心15min除杂质,取上清分装,每管70ul,—80oC保存。
2. Bradford法测蛋白含量取0.001g BSA(牛血清白蛋白)用1ml超纯水溶解,测定BSA标准曲线及样品蛋白含量。
取7个10ml的离心管,首先在5个离心管中按次序加入0ul, 5ul,10ul, 15ul, 20ul 的BSA溶解液,另2管中分别加入2 ul的待测样品溶液,再在每管中加入相应体积的双蒸水(总体积为80ul),然后,各管中分别加入4ml的Bradford液(原来配好的Bradford液使用前需再取需要的剂量过滤一遍方能使用),摇匀,2min在595nm下,按由低到高的浓度顺序测定各浓度BSA的OD值,再测样品OD值。
(测量过程要在一个小时内完成)。
3. 双向电泳第一向---IEF(双向电泳中一律使用超纯水)3.1 水化液的制备称取2.0mg 的DTT,用700ul水化液储液溶解后,加入8ul 0.05, 的溴酚兰,3.5ul(0.5,v/v)IPG buffer (pH 3-10)振荡混匀,13200rpm离心15min 除杂质,取上清。
在含300ug 蛋白(经验值)的样品溶解液中加入水化液,至终体积为340ul,振荡器上振荡混合,13200rpm离心15min除杂质,取上清。
3.2 点样,上胶分两次吸取样品,每次170ul, 按从正极到负极的顺序加入点样槽两侧,再用镊子拨开 Immobiline DryStrip gels (18cm,pH 3—10)胶条,从正极到负极将胶条压入槽中,胶面接触加入的样品。
双向电泳泳实验方案

双向电泳实验方案1.双向电泳总蛋白(培养细胞)提取方法:方案一细胞培养在10cm的培养皿中,待培养至80%左右密度时,细胞用预冷的PBS漂洗3次,加450μl裂解液,细胞刮刀收集,用移液器转移至1.5ml离心管中,反复吹打。
(折合在六孔板中,每孔加裂解液50μl)之后将样品用超声波细胞破碎仪超声处理超声时间为5s,间歇时间为10s,功率为100-120W,超声处理至溶液清澈无粘稠物为止,处理过程在冰浴中进行,超声处理后,4℃、25000g离心1h。
取上清进行蛋白质浓度测量,按实验所需的量分装后-80℃冰箱中保存。
(裂解液成分:8mol/L脲,65mmol/L DDT,4%CHAPS,40mmol/L Tris)方案二1.1试剂(1)抽提缓冲液9mol/L脲 5.4g4%CHAPS 0.4g0.5%IPG缓冲液(PH 3-10)(AP Biotech) 50.0μl50mmol/L DDT 0.077gH2O 加至10ml(2)IPG缓冲液(PH 3-10)(AP Biotech)(3) 磷酸盐缓冲液(PBS),冰冻1.2仪器(1) 细胞刮刀(2)离心机(低温,低速)(3)滤纸(4)冰浴装置(5)超速离心机(低温)(6)漩涡混合器1.3细胞培养的GC-1 spg细胞1.4方法1.从培养皿中转移细胞:用细胞刮刀从培养皿中刮下细胞,用5ml移液器将培养基和细胞转移至15ml离心管中。
2.480g、4℃离心沉淀细胞5min。
3.弃去上清,勿搅动沉淀要点:操作一下步骤时,所有细胞需保持冰冻状态;不离心或震荡时保持细胞在冰上。
4.离心管中加入10ml冰冻PBS,来回吹打重悬细胞。
5.480g、4℃再次离心细胞5min。
6.弃去上清,勿搅动沉淀。
7.重复步骤4-6两次。
8.在最后一次洗涤后,把离心管完全空干,用滤纸将沉淀上残留的PBS吸干。
9.用移液器将抽提缓冲液加到离心管中。
依赖所研究的细胞系来确定抽提缓冲液的体积。
双向电泳操作步骤

3.1 全菌蛋白的制备将细菌接种于DMEM培养基,置于28℃培养箱培养18h。
参照Coelho 等(Coelho et al. 2004)的方法,略有改动。
用Wash buffer(10mmol/L TrisCl pH8.0,5mmol/L 醋酸镁)清洗细胞3次。
离心,细胞沉淀在Lysis Buffer( 7mol/L 尿素,2mol/L硫尿,1% IPG Buffer pH3-10或pH4-7,4% CHAPS,1% DTT,1%蛋白酶抑制剂,1%核酶抑制剂)中悬浮,使其浓度范围在5~10mg/mL。
置于冰上裂解2h。
13000r/min离心1h取上清,-80℃保存。
用2-D clean-up Kit 纯化蛋白,再用2-D Quant Kit测定样本中蛋白浓度。
3. 2 2-D Clean-Up Kit的使用方法(全程小心)蛋白质样本在1.5mL微型离心管中处理,所有步骤均在冰上进行。
1)将体积100μL的蛋白质样本(含1~100μg蛋白质)置于1.5mL微型离心管中。
加入300μL沉淀剂。
振荡或倒置搅匀。
冰浴中(4~5℃)培育15min。
2)加入300μL共沉淀剂,简单振荡混合一下,12000r/min离心5min。
3)将上清液尽量多地倾析或吸出,不要搅散沉淀。
保持沉淀不变,在其上面加入一层40μL共沉淀剂,冰上培育5min。
后离心5min,去上清。
4)往沉淀加入25μL蒸馏水或去离子水。
将管振荡5~10s,这时沉淀应散开,但并未溶解于水中。
在管中加入1mL洗涤缓冲液(在-20℃下至少预冷1h)和5μL洗涤添加剂,振荡直至沉淀完全散开。
-20℃下培育至少30min,每10min振荡20~30s。
5)将离心管以最大速度(至少12000r/min)离心5min。
小心地将上清液移走弃去。
此时应可见白色沉淀,将沉淀简单风干一下(不要超过5min)。
6)用裂解液溶解沉淀,以备第一向IEF电泳。
振荡管子至少30s,在室温下孵育,振荡或用移液管抽吸使之完全溶解。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
细胞裂解(蛋白质提取)一、试剂配置:PBS配方氯化钠(MW 58.44) 130mM 8g氯化钾(MW 74.5) 2.7mM 0.2g十二水和磷酸氢二钠(MW 358) 10 mM3.63g磷酸二氢钾(MW 136) 2 mM0.24g加水至1L配好后进行高压灭菌。
Washing buffer(500mL)配方Tris(MW 121.1) 10mM 0.605g蔗糖(MW 342) 250mM 42.75g加水溶解,用HCl调pH至7.0后用孔径为0.22µm滤膜过滤(使用1M盐酸略高于2750uL调pH)裂解储液配方(细胞):尿素(MW 60.06) 7M 4.2g硫脲(MW 76.12) 2M 1.52gCHAPS(MW614.89) 4%(W/V)0.4g 加超纯水定容至10mL后经0.22µm一次性滤膜过滤,过滤后分装成20小管,每小管500uL,-20℃冰箱保存。
裂解储液配方(组织):尿素(MW 60.06)5M3g硫脲(MW 76.12) 2M1.52gCHAPS(MW614.89) 2% 0.2gTris(MW 121.1)40mM 0.048g加超纯水定容至10mL后经0.22µm一次性滤膜过滤,过滤后分装成20小管,每小管500uL,-20℃冰箱保存。
裂解液:裂解液储液 100uLIPG buffer(pH可选) 2% 2uLPi 2uLNucLease mix(100×) 1uLPMSF(100mM:20mg/mL) 1mM 1uLDTT(0.411g/mL) 40mM 1.5uLPi:每片使用200uL超纯水溶解后按10 uL分装考马斯亮蓝G-250:考马斯亮蓝G-250 0.01% 100g95%乙醇 4.7% 50mlH3PO4 8.5% 85g 将考马斯亮蓝G-250溶于50ml95%乙醇中,与用水溶解的100mlH3PO4混合后稀释至1000ml,之后使用滤纸过滤。
二、实验准备:1、准备冰盒,细胞裂解过程均在冰盒内进行(4℃);2、准备4℃离心机,开机降温,保证温度下降至4℃;3、取出置于-20℃保存的试剂,需冰上融化,最后取出细胞样品。
三、样品制备:(一)细胞细胞裂解准备:1、收集细胞,使用大于500g转速离心5min;2、弃去上清,使用washing buffer重悬细胞沉淀,震摇待细胞完全打散后,继续用大于500g转速离心5min;3、重复步骤2两次,并使用转速2000rpm离心5min,弃上清。
细胞可置于-80冰箱保存数周,或使用裂解液裂解。
细胞裂解:1、配置裂解液:注意先不要加入DTT。
临用前加入PMSF后,立即将细胞裂解液加入待裂解的细胞中。
2、加入裂解液的细胞置于冰盒内,每隔5-7min取出,于漩涡振荡器上震摇数秒,并再次放于冰盒中,保证细胞裂解时的温度为4℃。
3、裂解15-20min后加入DTT。
4、裂解完成后,置于4℃离心机中,以最大转速13200rpm离心30min,取上清,弃沉淀。
上清所含提取的蛋白。
5、使用bradford法测定提取蛋白浓度。
(二)、组织:组织裂解准备:1、提取动物组织。
提取时务必保证组织的纯度,尽量减少组织混杂,尤其注意血液的混入。
2、使用washing buffer冲洗组织2-3遍,洗净组织的血液、体液、和其他影响实验结果的残留成分。
3、准备研钵,洗净并且烘干,准备适量的组织裂解液,准备充足的液氮,石英砂适量。
组织裂解:1、配置裂解液:注意先不要加入DTT、 PMSF。
2、将适量石英砂加入到研钵中,加入液氮冷却。
3、将组织放入装有液氮的研钵中,研磨组织,在研磨过程中保证样品在低温状态下。
4、研磨完成后将样品及石英砂收集到离心管中,在裂解液中迅速加入PMSF,混匀后将裂解液加入含有样品的离心管中。
保持4℃,震荡混匀,每隔5-7min取出,于漩涡振荡器上震摇数秒,并再次放于冰盒中。
5、裂解15-20min后加入DTT。
6、使用2000g离心10min,吸取上清即为所提取的蛋白,下层石英砂及组织碎片则丢弃。
将上清使用13200g转速离心,弃去沉淀。
(三)、血清:血清样品可直接用于双向电泳实验。
但样品中含有大量的IgG和白蛋白,需要使用试剂盒去除。
四、试剂说明:裂解液成分:溶液中还原剂的含量不要超过20mM。
尿素/硫脲:变性剂,中性促溶剂,促进样品溶解,破坏氢键等次级结构,使所有离子基团暴露在溶液里。
硫脲可以促进膜蛋白的溶解。
尿素在30℃以上容易降解,降解产物异氰酸盐可使样本中蛋白质甲氨酰化,对蛋白质进行修饰,造成等电点改变,形成人为地“斑点串”。
CHAPS:两性表面活性剂,CHAPS是一种非变性的zwitterionic 去垢剂、蛋白质裂解液,用于增溶膜蛋白和裂解蛋白-蛋白之间的相互作用。
CHAPS是两性离子去垢剂,分子结构中具有一个胆酸和硫代甜菜碱。
在紫外区的低吸光性成为用紫外方法检测膜蛋白的首选去垢剂,CMC值均为8 mM。
常用于于双向电泳等实验。
原理:CHAPS保护蛋白质的天然状态,可溶解膜蛋白,从而互作用。
透析可除去。
SDS:(FW 288.38)离子型去垢剂,裂解力强,基本可以把细胞完全破坏掉。
并且使蛋白变性失活。
不得与强氧化剂混合。
它也可以用于核酸抽提操作中破坏细胞壁及裂解核酸蛋白复合物。
NP-40:乙基苯基聚乙二醇(Nonidet P40;)很温和的非离子型去垢剂,1%浓度的基本可以破坏掉胞膜,而对核膜破坏的作用弱,结合特定的buffer可以获得胞浆蛋白。
与蛋白结合力强,用于防止物质分子疏水间相互作用,确保蛋白的充分溶解和结构稳定。
尤其用于膜蛋白的非变性条件下的溶解。
TritonX-100:聚乙二醇辛基苯基醚,一种非离子型表面活性剂,能溶解脂质,以增加抗体对细胞膜的通透性。
1%的Triton X-100常用于漂洗组织标本,0.3%的Triton X-100则常用于稀释血清,配制BSA等。
能力介于NP-40和SDS之间,偏向于NP-40,在保护蛋白活性方面有一定作用。
EDTA:变性剂和稳定剂,通过螯合金属离子有效抑制蛋白酶的活性。
DTT:二硫苏糖醇,还原剂,储液配置:0.4g/mL。
使用浓度约为40mM。
防止蛋白通过巯基氧化聚合,增加蛋白的可溶性。
DTE:二硫赤酰糖醇,与DTT近似,也做还原剂。
ASB14: Amidosulfobetaine 14,TBP:tri-Butyl-Phosphate磷酸三丁酯、三丁基膦,有毒,具有较低的表面张力,难溶于水,常用作消泡剂。
非巯基还原剂,使用浓度为2mM,但溶解性有限,在溶液中不稳定,在样品制备时需DTT联合使用。
脱氧胆酸钠:Sodium deoxycholate,中度变性剂和蛋白溶解剂。
SB3-10:硫代甜菜碱10,诱食剂,甲基供给剂。
DeStreak试剂和DeStreak水化溶液:将蛋白质巯基转变为稳定地二硫化物基团,防止发生非特异性氧化反应,有效减少蛋白质在电泳过程中因再氧化而发生的拖尾,特别是pH7-11的范围内(碱性蛋白质)。
PMSF:苯甲基磺酰氟,储液配置(100mM):20mg/mL,在水中易失活,使用异丙醇或DMSO配置。
强大的不可逆蛋白酶抑制剂,抑制丝氨酸水解酶和一些半胱氨酸水解酶以及含有巯基的蛋白酶,使用浓度为1mM。
浓度测定:核酸酶(Nuclease mix):核酸酶的构成是由牛胰腺Dnase 和Rnase以及维持最佳核酸酶活性所必须的各种辅助因子组成。
核酸酶受EDTA的干扰,使用核酸酶时避免加入EDTA。
可与蛋白酶抑制剂(PI)同时使用,但要注意必须选择无EDTA的蛋白酶抑制剂。
(注意:核酸酶试剂组为悬浮液,使用前务必震荡混匀后分装。
)考马斯亮蓝:Coomassie brilliant blue.考马斯亮蓝G-250(Coomassie brilliant blue G-250)比考马斯亮蓝R250多二个甲基。
MW=854;λmax=590―610nm。
测定蛋白质含量属于染料结合法的一种。
考马斯亮蓝G-250在游离状态下呈红色,最大光吸收在488nm;当它与蛋白质结合后变为青色,蛋白质-色素结合物在595nm波长下有最大光吸收。
其光吸收值与蛋白质含量成正比,因此可用于蛋白质的定量测定。
蛋白质与考马斯亮蓝G-250结合在2min左右的时间内达到平衡,完成反应十分迅速;其结合物在室温下1h内保持稳定。
灵敏度比Lowry 法还高4倍,染色灵敏度不如R250,但比氨基黑高3倍。
优点在于它在三氯乙酸中不溶而成胶体,能选择地染色蛋白而几乎无本底色。
所以常用于需要重复性好和稳定的染色,适于作定量分析。
可测定微克级蛋白质含量,测定蛋白质浓度范围为0~1 000μg/mL。
该方法用于大多数蛋白质的定量是比较精确的,但不适用于小分子碱性多肽的定量。
去污剂的浓度超过0.2%影响测定结果。
如TritonX-100、SDS、NP-40等。
考马斯亮蓝和皮肤中蛋白质通过范德华力结合,反应快速,并且稳定,无法用普通试剂洗掉。
待一两周左右,皮屑细胞自然衰老脱落即可无碍。
考马斯亮蓝R250与蛋白质反应虽然比较缓慢,但是可以被洗脱下去,所以可以用来对电泳条带染色。
R250较敏感,做定量实验用,染胶效果较好.染色后为红蓝色。
染色灵敏度比氨基黑高5倍。
尤其适用于SDS电泳微量蛋白质染色。
但蛋白质浓度超出一定范围时,对高浓度蛋白的染色不符合Beer定律,用作定量分析时要注意这点。
此方法优点:干扰物质少,K+、Na+、Mg2+离子、Tris缓冲液、糖和蔗糖、甘油、巯基乙醇、EDTA等均不干扰此测定法。
此法的缺点是:(1)由于各种蛋白质中的精氨酸和芳香族氨基酸的含量不同,因此Bradford法用于不同蛋白质测定时有较大的偏差,在制作标准曲线时通常选用 g—球蛋白为标准蛋白质,以减少这方面的偏差。
(2)仍有一些物质干扰此法的测定,主要的干扰物质有:去污剂、 Triton X-100、十二烷基硫酸钠(SDS)和0.1N的NaOH。
(如同0.1N的酸干扰Lowary法一样)。
(3)标准曲线也有轻微的非线性,因而不能用Beer定律进行计算,而只能用标准曲线来测定未知蛋白质的浓度。
蛋白质纯化一、目的:蛋白质纯化的目的在于去除样品蛋白中的盐、糖以及一些大分子杂质,杂质的存在严重影响双向电泳的结果。
二、方法:本实验室蛋白质常用的除杂方法主要有丙酮沉淀、TCA-丙酮沉淀、2D-cLeanup-kit处理。
丙酮沉淀:加入3-4倍体积的-20℃保存的丙酮沉淀大于1小时。
沉淀结束后使用13200rpm转速离心10min,去上清,将ep管置于冰上晾干5min,后用水化液溶解。
TCA-丙酮沉淀:使用含有10%TCA的丙酮溶液(100mL丙酮加入10gTCA(三氯乙酸),也可加入0.07%的DTT),沉淀方法与丙酮沉淀类似。