中国药典分析方法汇总
2020年中国药典运用高效液相色谱法测定含量的品种

2020年我国药典是一部权威的药品标准,其中包含了大量的药品、原料药和药用辅料的规范。
药品的质量和安全性对于人们的生命健康至关重要,而药典中的含量测定方法是评价药品质量的重要手段之一。
而高效液相色谱法(High Performance Liquid Chromatography, HPLC)作为一种快速、准确的分析方法,在药典中得到了广泛的应用。
本文主要介绍了2020年我国药典中运用高效液相色谱法测定含量的品种。
一、什么是高效液相色谱法高效液相色谱法是一种利用溶液在固定相载体上进行分配平衡的色谱分析方法。
它利用流动相的不同速度使样品中的成分在固定相上分离出来,再通过检测器对各个成分进行检测和定量。
该方法具有分离效率高、灵敏度高、准确性高、重现性好等优点,因此被广泛应用于药品质量控制领域。
二、2020年我国药典中运用高效液相色谱法测定含量的品种1.对乙酰氨基酚片对乙酰氨基酚片是一种用于退烧和镇痛的常用药品,其含量测定需要高效液相色谱法进行。
根据2020年我国药典,对乙酰氨基酚片中对乙酰氨基酚的含量可以采用高效液相色谱法进行测定,并以此作为评价药物质量的重要指标。
2.阿莫西林胶囊阿莫西林是一种广谱抗生素,常用于治疗呼吸道、泌尿生殖系统和消化道感染等疾病。
2020年我国药典规定,阿莫西林胶囊中阿莫西林的含量可以采用高效液相色谱法进行测定,以确保药品的质量。
3.复方甘草酸苷片复方甘草酸苷片是一种常用的抗炎、抗过敏药品,其含量测定也需要采用高效液相色谱法进行。
2020年我国药典规定,复方甘草酸苷片中甘草酸二钾的含量可以采用高效液相色谱法进行测定,并以此作为评价药物质量的重要指标。
4.硫酸链霉素滴眼液硫酸链霉素滴眼液是一种用于治疗眼部感染的药品,其含量测定也需要采用高效液相色谱法进行。
2020年我国药典规定,硫酸链霉素滴眼液中链霉素的含量可以采用高效液相色谱法进行测定,以确保药品的质量。
5.丙溴定胶囊丙溴定是一种中枢神经系统调节药,常用于治疗癫痫等疾病。
药物分析考点总结

名词术语含义避光系指用不透光的容器包装,例如棕色容器或黑纸包裹的无色透明、半透明容器密闭系指将容器密闭,以防止尘土及异物进入密封系指将容器密封以防止风化、潮解、挥发或异物进入熔封或严封阴凉处凉暗处系指将容器熔封或用适宜的材料严封,以防止空气与水分的侵入并防止污染系指不超过20℃系指避光并不超过20℃第一章药典1.国家药品标准包括:《中国药典》、《药品标准》、药品注册标准。
2.药品标准的制定原则(1)检测项目的制定要有针对性(2)检验方法的选择要有科学性(3)限度规定的规定要有合理性3.《中国药典》,缩写为Ch.P。
我国现已出版了九版药典。
现在每五年制定一次。
4.组成:一部、二部、三部及其增补本。
第一部收载中药材及饮片,植物油脂和提取物,成方制剂和单味制剂。
第二部收载化学药品、抗生素、生化药品、放射性药品及其制剂及药用辅料。
第三部收载生物制品。
5.《中国药典》内容:凡例、正文和附录6.“凡例”是为正确使用《中国药典》进行药品质量检定的基本原则,是对《中国药典》正文、附录及与质量检定有关的共性问题的统一规定。
7.《中国药典》正文收载的中文药品名称系按照《中国药品通用名称》收载的名称及其命名原则命名,为药品的法定名称。
英文名均采用国际非专利药名(INN)。
8.名称单位长度体积质量压力动力黏度运动黏度波数密度放射性活度m, dm, cm, mm, μm, nm L,ml,μlKg, g, mg, μg,ng Mpa, kPa, PaPa.s,mPa.sm2/s mm 2/scm-1kg/m 3 g/cm 3GBq MBq kBq Bq9.原料药的含量(%),除另有注明者,均按重量计。
如规定上限为100%以上时,系指用药典规定的分析方法测定时可能达到的数值,它为药典规定的限度或允许偏差,并非真是含量;如未规定上限时,系指不超过101.0%。
10.标准品、对照品系指用于鉴别、检查、含量测定的标准物质。
由国务院药品监督管理部门指定的单位制备、标定和供应。
《中国药典》2015版四部9101药品质量标准分析方法验证指导原则

《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则《中国药典》2015版四部9101药品质量标准分析⽅法验证指导原则药品质量标准分析⽅法验证的⽬的是证明采⽤的⽅法适合于相应检测要求。
在建⽴药品质量标准时,分析⽅法需经验证;在药品⽣产⼯艺变更、制剂的组分变更、原分析⽅法进⾏修订时,则质量标准分析⽅法也需进⾏验证。
⽅法验证理由、过程和结果均应记载在药品质量标准起草说明或修订说明中。
⽣物制品质量控制中采⽤的⽅法包括理化分析⽅法和⽣物学测定⽅法,其中理化分析⽅法的验证原则与化学药品基本相同,所以可参照本指导原则进⾏,但在进⾏具体验证时还需要结合⽣物制品的特点考虑;相对于理化分析⽅法⽽⾔,⽣物学测定⽅法存在更多的影响因素,因此本指导原则不涉及⽣物学测定⽅法验证的内容。
验证的分析项⽬有:鉴别试验、限量或定量检查、原料药或制剂中有效成分含量测定,以及制剂中其他成分(如防腐剂等,中药中其他残留物、添加剂等)的测定。
药品溶出度、释放度等检查中,其溶出量等的测定⽅法也应进⾏必要验证。
验证指标有:准确度、精密度(包括重复性、中间精密度和重现性)、专属性、检测限、定量限、线性、范围和耐⽤性。
在分析⽅法验证中,须采⽤标准物质进⾏试验。
由于分析⽅法具有各⾃的特点,并随分析对象⽽变化,因此需要视具体⽅法拟订验证的指标。
表1中列出的分析项⽬和相应的验证指标可供参考。
表1检验项⽬和验证指标项⽬鉴别杂质测定含量测定及校正因⼦内容定量限度溶出量测定准确度-+-++精密度重复性-+-++中间精密度-+①-+①+专属性②+++++检测限--③+--定量限-+--+线性-+-++范围-+-++耐⽤性+++++①巳有重现性验证,不需验证中间精密度。
②如⼀种⽅法不够专属,可⽤其他分析⽅法予以补充。
③视具体情况予以验证。
⼀、准确度准确度系指采⽤该⽅法测定的结果与真实值或参考值接近的程度,⼀般⽤回收率(%)表⽰。
准确度应在规定的范围内测定。
9101药品质量标准分析方法验证指导原则-15版药典

回收率 % = ( C _ A ) /S X 100 % 式 中 A 为供试品所含被测成分量;
B 为加入对照品量; C 为实测值。 4 . 校正因子的准确度 对 色 谱 方 法 而 言 ,绝 对 (或 定 量 )校 正 因 子 是 指 单 位 面 积 的 色 谱 峰 代 表 的待 测 物 质 的 量。待测定物质与所选 定的参照 物 质 的绝 对校 正 因子 之 比 ,即 为 相 对 校 正 因 子 。相对校正因 子计算法常应用于化学药有关物质的测定、中药材及其复方 制剂中 多指标 成分 的 测 定 。校 正 因 子 的 表 示 方 法 很 多 ,本指 导原则中的校正因子是指气相色谱法和髙效液相色谱法中的 相对重量校正因子。 相 对 校 正 因 子 可 采 用 替 代 物 (对 照 品 )和 被 替 代 物 (待测 物) 标 准 曲 线 斜 率 比 值 进 行 比 较 获 得 ;采 用 紫 外 吸 收 检 测 器 时 ,可 将 替 代 物 (对 照 品 )和 被 替 代 物 (待 测 物 )在 规 定 波 长 和 溶 剂 条 件 下 的 吸 收 系 数 比 值 进 行 比 较 ,计 算 获 得 。 5 . 数据要求 在 规 定 范 围 内 ,取 同 一 浓 度 (相 当 于 100%浓 度 水 平 )的 供 试 品 ,用 至 少 测 定 6 份 样 品 的 结 果 进 行 评 价 ;或 设 计 3 种 不 同 浓 度 ,每 种 浓 度 分 别 制 备 3 份 供 试 品 溶 液 进 行 测 定 ,用 9 份 样 品 的 测 定 结 果 进 行 评 价 。对 于 化 学 药 ,一 般 中 间 浓 度 加人量与所取供试品中待测定成分量之比控制在1 : 1 左右,
中国药典分析检测方法技术指南

中国药典分析检测方法技术指南下载温馨提示:该文档是我店铺精心编制而成,希望大家下载以后,能够帮助大家解决实际的问题。
文档下载后可定制随意修改,请根据实际需要进行相应的调整和使用,谢谢!并且,本店铺为大家提供各种各样类型的实用资料,如教育随笔、日记赏析、句子摘抄、古诗大全、经典美文、话题作文、工作总结、词语解析、文案摘录、其他资料等等,如想了解不同资料格式和写法,敬请关注!Download tips: This document is carefully compiled by the editor. I hope that after you download them, they can help yousolve practical problems. The document can be customized and modified after downloading, please adjust and use it according to actual needs, thank you!In addition, our shop provides you with various types of practical materials, such as educational essays, diary appreciation, sentence excerpts, ancient poems, classic articles, topic composition, work summary, word parsing, copy excerpts,other materials and so on, want to know different data formats and writing methods, please pay attention!中国药典分析检测方法技术指南是一部重要的医药行业规范性文献,它为药物质量控制和安全性评价提供了重要支持。
药典常用定量分析方法概述

分 析 化 学
酸碱滴定(非水酸碱滴定法) 配位滴定 容量分析法 沉淀滴定 氧化还原滴定 纸色谱PC 电化学分析法 平面色谱 经典色谱 薄层色谱TLC ☆色谱分析 柱色谱法(CC) 气相色谱法(GC) 现代色谱 *高效液相色谱法(HPLC) 高效毛细管电泳法(HPCE) 紫外—可见分光光度法(UV—Vis) 红外分光光度法IR 原子吸收分光光度法(AAS) 光谱分析 原子发射分光光度法(AES) 或波谱分析 荧光分析法(Fluor) 核磁共振法(NMR) 质谱法(MS)
(三)碘量法误差的主要来源
1.碘的挥发 预防: 1)过量加入KI——助溶,防止挥发 增大浓度,提高速度 2)溶液温度勿高 3)碘量瓶中进行反应(磨口塞,封水) 4)滴定中勿过分振摇 2.碘离子的氧化(酸性条件下) 预防: 1)控制溶液酸度(勿高) 2)避免光照(暗处放置) 3)I2完全析出后立即滴定 4)除去催化性杂质(NO3-,NO,Cu2+)
注:无须知道CI2
置换碘量法测定CuSO4的含量
2Cu2+ + 4I- (过量) 2CuI ↓ + I2 I2 + 2S2O322I- + S4O62-
CuSO4 %
(C Na2 S 2O3 VNa2 S 2O3 ) M CuSO4 S 1000
100 %
注: • CuI易水解,故以HAc为介质 • CuI强烈吸附I2造成终点提前,滴定时应用力振摇 或加入KSCN转化CuI沉淀为CuSCN,同时释放 I2
实质:电子的转移 特点: 1)机理复杂、多步反应 2)有的程度虽高但速度缓慢 3)有的伴有副反应而无明确计量关系 分类: 碘量法、高锰酸钾法、重铬酸钾法、 亚硝酸钠法、溴量法、铈量法 应用:广泛,直接或间接测定无机物、有机物
中国药典2020年版 9101 分析方法验证指导原则
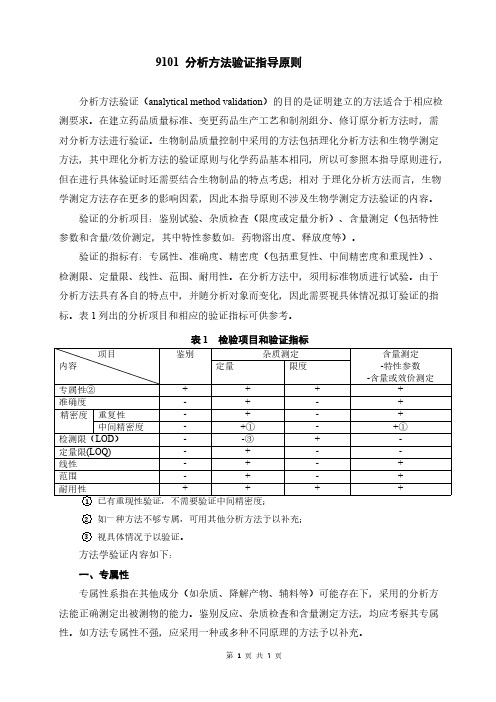
9101分析方法验证指导原则分析方法验证(analytical method validation)的目的是证明建立的方法适合于相应检测要求。
在建立药品质量标准、变更药品生产工艺和制剂组分、修订原分析方法时,需对分析方法进行验证。
生物制品质量控制中采用的方法包括理化分析方法和生物学测定方法,其中理化分析方法的验证原则与化学药品基本相同,所以可参照本指导原则进行,但在进行具体验证时还需要结合生物制品的特点考虑;相对于理化分析方法而言,生物学测定方法存在更多的影响因素,因此本指导原则不涉及生物学测定方法验证的内容。
验证的分析项目:鉴别试验、杂质检查(限度或定量分析)、含量测定(包括特性参数和含量/效价测定,其中特性参数如:药物溶出度、释放度等)。
验证的指标有:专属性、准确度、精密度(包括重复性、中间精密度和重现性)、检测限、定量限、线性、范围、耐用性。
在分析方法中,须用标准物质进行试验。
由于分析方法具有各自的特点中,并随分析对象而变化,因此需要视具体情况拟订验证的指标。
表1列出的分析项目和相应的验证指标可供参考。
表1检验项目和验证指标项目内容鉴别杂质测定含量测定-特性参数-含量或效价测定定量限度专属性②++++准确度-+-+精密度重复性-+-+中间精密度-+①-+①检测限(LOD)--③+-定量限(LOQ)-+--线性-+-+范围-+-+耐用性++++ 1已有重现性验证,不需要验证中间精密度;2如一种方法不够专属,可用其他分析方法予以补充;3视具体情况予以验证。
方法学验证内容如下:一、专属性专属性系指在其他成分(如杂质、降解产物、辅料等)可能存在下,采用的分析方法能正确测定出被测物的能力。
鉴别反应、杂质检査和含量测定方法,均应考察其专属性。
如方法专属性不强,应采用一种或多种不同原理的方法予以补充。
1.鉴别反应应能区分可能共存的物质或结构相似的化合物。
不含被测成分的供试品,以及结构相似或组分中的有关化合物,应均呈阴性反应。
药物分析-中国药典

药典概况《中华人民共和国药品管理法》(2019年修订)中规定,国务院药品监督管理部门颁布的《中华人民共和国药典》(以下简称“《中国药典》”)和药品标准为国家药品标准。
国家药品标准是国家为保证药品质量,对药品的质量指标、检验方法等作出的强制性技术规定。
《中国药典》是国家药品标准体系的核心,是药品生产经营者的基本遵循,是药品监督管理工作的准绳。
药典是收载国家药品标准的法典。
我国的药典为《中华人民共和国药典》,简称《中国药典》,其英文名称是Pharmacopoeia of The People’s Republic of China,简称Chinese颁布实施。
《中国药典》的沿革与进展一、ChP的历史沿革1953年版:第一版, 共收载531种;1963年版:第二版, 共收载1310种; 一部:中药材和中药成方制剂二部:化学药品。
1977年版:第三版,共收载1925种;一部:中草药、提取物、植物油脂、中药成方制剂;二部:化学药品、生物制品。
1985年版:第四版,共收载1489种;一部:中草药、提取物、植物油脂、中药成方制剂;二部:化学药品、生物制品。
1990年版:第五版,共收载1751种;一部:中草药、提取物、植物油脂、中药成方制剂;二部:化学药品、生物制品;《药品红外光谱集》出版。
1995年版:第六版,共收载2375种;一部:中草药、提取物、植物油脂、中药成方制剂;二部:化学药、抗生素、生化药、放射性药物、生物制品及辅料;《临床用药须知》、《药品红外光谱集》、《中药彩色图集》、《中国药品通用名称》出版。
2000年版:第七版,共收载2691种;一部:中草药、提取物、植物油脂、中药成方制剂;二部:化学药品、生物制品;首次药品标准分析方法验证要求等六项指导原则。
2005年版:第八版,共收载3217种。
分为一部、二部、三部,首次将《中国生物制品规程》纳入其中,并单独列为《中国药典》三部;附录采用原子吸收或电感耦合等离子体质谱法测定重金属和有害金属。