基于 Orbitrap GC-MS 的非靶向代谢组学
GC-MS非靶标代谢组分析

百泰派克生物科技
GC-MS非靶标代谢组分析
代谢组学是一种全面的代谢分析方法,能以无偏见的方式同时分析广泛的代谢物类别。
代谢组学方法可分为靶向和非靶向两种。
靶向代谢组学是指对一组选定的代谢物(例如氨基酸、脂质、糖和/或脂肪酸)进行定量测量,以研究特定的代谢途径
或验证使用非靶向代谢分析确定的生物标志物。
相比之下,非靶向代谢组学方法涉及代谢组的全局分析,旨在快速可靠地识别特定生理状态的小分子生物标志物,与靶向代谢组学相比,非靶向代谢组学通常可提供更多信息。
GC-MS非靶标代谢组学通过气相色谱-质谱联用技术对生物体液、组织和细胞中的
所有小分子代谢物进行综合分析,检测实验组和对照组中所有小分子代谢物的动态变化,旨在寻找生物体受外界刺激前后体内有显著变化的代谢物,揭示这些代谢物与生理病变之间的关系,为了解各种生物过程的信号通路以及小分子代谢物的调节作用提供了理论依据,广泛用于识别癌症生物标志物和调节肿瘤进展的代谢物研究。
百泰派克生物科技基于Thermo公司的Q Exactive和AB公司的Q TOF 5600质谱平台,推出GC/MS非靶向代谢组学分析服务技术包裹,基于之前发表的文献建立了多种样品处理的技术平台,您只需要将您的实验目的告诉我们并将您的样品寄给我们,我们会负责项目后续所有事宜,包括样品准处理、质谱分析、质谱原始数据分析、生物信息学分析。
uplc-ms的非靶向代谢组学方法

非靶向代谢组学方法是一种用于发现并分析生物体内所有代谢产物的方法。
其中,uplc-ms(超高效液相色谱-质谱联用)技术被广泛应用于非靶向代谢组学研究中,因其高灵敏度、高分辨率和高通量的特点而备受青睐。
本文将重点介绍uplc-ms的非靶向代谢组学方法,包括样品处理、色谱分离、质谱检测、数据处理等各个方面。
1. 样品处理在非靶向代谢组学研究中,样品处理是非常关键的一步。
经典的样品处理方法包括蛋白沉淀、溶剂提取和衍生化等。
对于不同类型的生物样品,比如血浆、尿液、组织、细胞等,都需要选择合适的样品处理方法来提取代谢产物。
2. 色谱分离uplc-ms技术的另一个关键步骤是色谱分离。
通过超高效液相色谱技术,可以对样品中的代谢产物进行高效、快速的分离。
色谱柱的选择、流动相的配制、梯度 elution等因素都会影响色谱分离的效果,因此需要进行精心的设计和优化。
3. 质谱检测uplc-ms技术的核心是质谱检测。
通过质谱仪器的高灵敏度、高分辨率和高质谱质量的特点,可以对样品中的代谢产物进行快速、准确的检测和分析。
质谱仪器的参数设置、离子扫描模式的选择、质谱图的解释等都是影响质谱检测结果的重要因素。
4. 数据处理完成了样品处理、色谱分离和质谱检测后,还需要对得到的海量数据进行处理和分析。
包括峰识别、质谱图的定量和定性分析、多变量统计分析等,都需要借助专业的数据分析软件和统计学方法来完成。
总结uplc-ms的非靶向代谢组学方法在生物医学、药物研发、环境科学等领域都有着广泛的应用前景。
通过精心设计各个步骤,结合先进的仪器设备和专业的数据处理技术,可以更全面、更深入地揭示生物体内代谢变化的规律,为疾病诊断、药物研发等提供有力支持。
希望uplc-ms的非靶向代谢组学方法在未来能够得到更广泛的推广和应用。
uplc-ms的非靶向代谢组学方法在生物医学、药物研发、环境科学等领域的广泛应用中,为科研工作者提供了强大的工具和技术支持。
非靶代谢组

非靶代谢组引言非靶代谢组(non-targeted metabolomics)是一种高通量的代谢组学技术,可以在不预先设定特定分子进行分析的情况下,综合地鉴定和定量生物体内的代谢物。
与靶向代谢组学不同,非靶代谢组学无需事先确定分析目标,而是通过大规模筛查样本中的代谢产物,以发现新的代谢通路和生物标志物。
本文将对非靶代谢组的原理、应用领域、技术流程和数据分析进行详细介绍,并讨论其在医学、生物学和食品科学等领域的潜在应用。
原理非靶代谢组学主要基于质谱(mass spectrometry)技术,结合色谱(chromatography)和核磁共振(nuclear magnetic resonance)等方法,实现对样本中代谢物的全面分析。
非靶代谢组学的分析流程包括样品制备、仪器检测和数据分析三个关键步骤。
首先,对样品进行适当处理,如提取、去蛋白和衍生化等,以增强代谢物的检测和分析能力。
然后,使用质谱等仪器对样品进行分析,将代谢物的质量-电荷比(m/z)和相对丰度信息记录下来。
最后,通过数据预处理、特征选择和统计分析等方法,对非靶代谢组学数据进行解析和解释。
应用领域非靶代谢组学在医学、生物学和食品科学等领域有着广泛的应用。
医学非靶代谢组学可以用于发现新的生物标志物,以辅助疾病诊断和预后评估。
通过比较病例组和正常对照组的代谢组学数据,可以鉴定与疾病相关的代谢异常,从而为疾病的早期诊断和治疗提供依据。
例如,非靶代谢组学已被应用于癌症、糖尿病和心血管疾病等多种疾病的研究中,帮助揭示其潜在的代谢机制。
生物学非靶代谢组学可以揭示生物体内代谢网络的复杂性和动态性,帮助解析代谢通路和代谢互作网络。
通过对不同生理状态下的代谢物进行分析,可以揭示代谢通路的变化规律,并研究其与生物学功能和调控的关联。
此外,非靶代谢组学还可以用于研究植物代谢物的合成、植物应答环境胁迫的机制等。
食品科学非靶代谢组学可以用于食品质量和安全的评估。
非靶向代谢组学,你想知道的都在这里
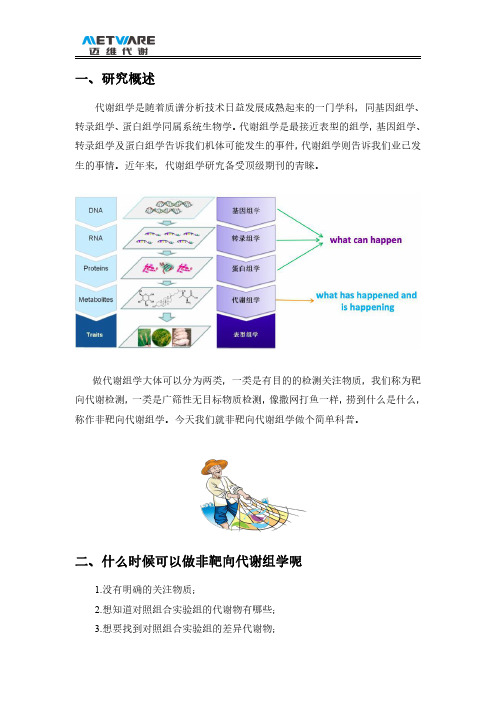
一、研究概述代谢组学是随着质谱分析技术日益发展成熟起来的一门学科,同基因组学、转录组学、蛋白组学同属系统生物学。
代谢组学是最接近表型的组学,基因组学、转录组学及蛋白组学告诉我们机体可能发生的事件,代谢组学则告诉我们业已发生的事情。
近年来,代谢组学研究备受顶级期刊的青睐。
做代谢组学大体可以分为两类,一类是有目的的检测关注物质,我们称为靶向代谢检测,一类是广筛性无目标物质检测,像撒网打鱼一样,捞到什么是什么,称作非靶向代谢组学。
今天我们就非靶向代谢组学做个简单科普。
二、什么时候可以做非靶向代谢组学呢1.没有明确的关注物质;2.想知道对照组合实验组的代谢物有哪些;3.想要找到对照组合实验组的差异代谢物;5.寻找疾病biomarker。
三、非靶向代谢组学的流程非靶向代谢组学使用的技术平台是LC-MS,用高效液相色谱作为分离系统,高分辨率质谱作为检测系统的一种串联分析平台。
其检测流程是是实验设计——样本收集——代谢物提取——代谢物检测——质谱数据采集——定性定量分析。
非靶流程如下图:四、非靶代谢组学做的好不好由什么决定非靶向代谢组学希望能够最大程度上体现生物样品体系中总的代谢物信息,筛选到的物质数越多越好;尽可能地去定性和定量(相对)所有的代谢物,准确度越高越好,这取决于多种因素,例如仪器、数据预处理及鉴定、生信分析手段。
仪器决定我们检测的灵敏度是多少、检测到物质峰有多少,数据库决定我们坚定出的物质是否准确、全面,生信分析手段决定我们面对从庞大的数据中获得想要的信息,怎样把我们找到的信息表达出来。
下面我们就从仪器、数据库、生信分析手段方法看一下如何做好非靶代谢组学。
仪器做代谢组学的仪器平台大体分为三种:核磁共振(NMR)、气质联用(GC-MS)、液质联用(LC-MS)。
它们各有优缺点,适合不同情境使用。
比如核磁共振可以做无创检测,如果你想检测活体小鼠的代谢情况,就可以选核磁共振,但是这种方法的灵敏度很低,不适合与灵敏度需求高的情境。
non-target_metabolomics_analysis_概述及解释说明

non-target metabolomics analysis 概述及解释说明1. 引言1.1 概述在生物学和医学领域中,代谢组学是一项重要的研究方法,旨在揭示生物体内代谢过程的整体状态以及与特定疾病之间的关联。
传统的代谢组学分析通常基于目标化合物检测,即预先选择需要检测的代谢产物进行定量分析。
然而,非目标代谢组学分析则更加全面地搜寻和识别样本中存在的所有代谢产物,从而将观察范围扩展到未知化合物和低丰度的代谢物。
1.2 文章结构本文将从以下几个方面对非目标代谢组学分析进行全面介绍:首先,在第2部分中我们会定义非目标代谢组学分析并探讨其技术原理;接着,在第3部分我们会详细阐述非目标代谢组学分析的步骤和方法,包括样本准备、色谱-质谱联用技术以及数据处理与分析平台;然后,在第4部分中我们会探讨非目标代谢组学分析的优势和挑战;最后,在第5部分我们将总结主要发现和贡献,并展望未来的研究方向和应用前景。
1.3 目的本文旨在提供关于非目标代谢组学分析的全面概述,以帮助读者了解该领域的基本原理、方法和应用。
同时,我们也希望通过探讨非目标代谢组学分析的优势和挑战,引起对该技术在生物医学研究中发展和应用的兴趣。
通过本文的阅读,读者将能够深入了解非目标代谢组学分析及其在揭示代谢调控机制和疾病诊断等方面的潜力。
2. 非目标代谢组学分析:2.1 定义:非目标代谢组学分析是一种无需预设目标分子的高通量代谢物筛选方法。
与传统的目标代谢分析相比,非目标代谢组学分析可以全面地检测样品中的代谢产物,并帮助我们了解生物系统在不同条件下的整体代谢状态。
2.2 技术原理:非目标代谢组学分析主要依靠色谱-质谱联用技术(LC-MS/MS)进行实验。
首先,通过样品准备步骤,包括样品提取和预处理,将样品中的代谢产物提取出来并准备好进行分析。
接下来,将提取出的化合物通过液相色谱(LC)进行分离,并将其引入质谱仪进行检测和质量鉴定。
最后,利用数据库以及专门开发的数据处理与分析平台对获得的数据进行解析和解释。
非靶和靶向 代谢组学;转录;蛋白组学;宏基因组学

非靶和靶向代谢组学、转录组学、蛋白组学和宏基因组学是近年来在生物医学领域中备受关注的研究方向。
这些研究方法和技术不仅为生命科学领域的研究提供了新的视角和手段,也为疾病的筛查、诊断和治疗提供了新的思路和途径。
本文将分别就这四个研究领域进行介绍,分析其在生物医学领域中的应用和发展前景。
一、非靶和靶向代谢组学1. 非靶代谢组学是指在没有预设代谢产物的假设下,全面分析生物样本中的所有代谢产物。
该技术通过质谱和核磁共振等方法对生物样本中的代谢产物进行检测和分析,从而揭示生物体内的代谢组成和代谢途径。
非靶代谢组学已被广泛应用于疾病的早期诊断、疾病机制的研究和药物的研发等领域,展现出了巨大的应用潜力。
2. 靶向代谢组学则是一种有目的地筛选和分析特定代谢产物的方法。
通过这种技术,研究者可以有针对性地对某些代谢物进行深入研究,从而更好地理解其在疾病发生发展中的作用机制。
靶向代谢组学在肿瘤研究、心血管疾病研究等方面取得了重要进展。
二、转录组学1. 转录组学是一种全面研究生物体内全部转录本的方法。
通过高通量测序技术,研究者可以获得生物样本中所有mRNA的序列信息,从而全面了解生物体内基因的表达情况和调控网络。
转录组学已被广泛应用于肿瘤、免疫系统疾病等领域,为疾病的诊断和治疗提供了重要参考。
2. 近年来,单细胞转录组学技术的发展为转录组学研究带来了新的突破。
该技术能够从单个细胞中获得转录组信息,揭示不同细胞类型和状态下的转录差异,为细胞分化、疾病发生发展等提供了重要线索。
三、蛋白组学1. 蛋白组学是一种全面研究生物体内全部蛋白质的方法。
通过质谱等技术,研究者可以了解生物样本中所有蛋白质的种类、表达水平和修饰情况,从而全面了解蛋白质在生物体内的功能和调控机制。
蛋白组学已被广泛应用于肿瘤标志物的发现、药物靶点的筛选等研究领域。
2. 磷酸化、甲基化等蛋白质修饰的研究成果为蛋白组学研究带来了新的发展方向。
研究者可以通过蛋白组学技术对这些蛋白质修饰进行深入研究,从而揭示它们在疾病发生发展中的作用机制。
非靶向代谢组学结果解读

非靶ห้องสมุดไป่ตู้代谢组学的结果解读主要包括以下几个方面:
1.代谢物识别:对非靶向代谢组学结果进行代谢物的识别和鉴定。这是分析代谢组学数据的首要步骤,需要使用质谱分析技术和数据库匹配等方法,确定代谢物的化学结构和分子式。
4.生物标志物筛选:利用差异分析和通路分析的结果,鉴定出具有临床应用潜力的生物标志物,为疾病的早期诊断和治疗提供有力支持。
总之,非靶向代谢组学结果的解读需要综合运用多种分析方法和技术手段,从不同层面和角度进行分析,从而揭示代谢物变化的生物学意义。
2.差异分析:比较不同样本之间代谢物的含量差异,找出具有显著性差异的代谢物,以及这些代谢物的生物学功能和代谢途径。通过这种方式可以发现不同生理状态或疾病状态下代谢物的变化规律,为进一步的研究提供线索。
3.通路分析:将代谢物按照代谢途径进行分类,分析不同代谢途径的变化趋势,以及这些代谢途径与生物学过程的关系。通过这种方式可以揭示代谢物变化背后的生物学机制,为进一步的研究提供理论基础。
代谢组学非靶向物质鉴定

代谢组学非靶向物质鉴定
代谢组学是一种对生物体内代谢产物进行系统分析的技术,其中包括对非靶向物质的鉴定。
非靶向代谢组学是一种不针对特定代谢产物进行分析的方法,它可以检测到生物体内所有的代谢产物,并对其进行定性和定量分析。
在非靶向代谢组学中,代谢产物的鉴定是一个关键步骤。
通常,代谢产物的鉴定是通过将未知代谢产物的质谱数据与已知代谢产物的数据库进行比对来实现的。
这种方法称为“谱库检索”,它可以根据未知代谢产物的质谱数据与数据库中已知代谢产物的质谱数据进行比对,从而确定未知代谢产物的身份。
然而,由于生物体内代谢产物的数量庞大,且许多代谢产物的结构相似,因此单纯依靠谱库检索往往难以准确鉴定代谢产物。
因此,在非靶向代谢组学中,还需要结合其他技术来提高代谢产物的鉴定准确性。
其中一种常用的技术是“多级质谱”,它可以将代谢产物的质谱数据分解成多个碎片离子的质谱数据,从而提供更多的结构信息。
此外,还可以结合化学结构分析、同位素标记等技术来进一步提高代谢产物的鉴定准确性。
总之,非靶向代谢组学中的代谢产物鉴定是一个复杂的过程,需要结合多种技术和方法来提高准确性。
随着技术的不断发展和完善,代谢产物的鉴定准确性将会不断提高,为生物医学研究提供更有力的支持。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
高亮区域中峰强度随时间的变化。
BP: 218.10272 @ 5.44E+008
化合物发现阶段 Q Exactive GC 系统获取了每个样品的全扫描色谱图(图 2)。 该系统在较宽的动态范围内采集不同浓度水平代谢物的全扫描 色谱图,不会丢失任何精确质量数信息。使用自动峰选取(结 合使用 XCMS 和 MzMatch.R)功能提取每个 EI 峰簇。在该步 骤中,已检测到 1193 个明显峰簇并进行了初步定量,强度阈 值为 100,000。已解卷积峰簇的示例见图 3。
仪器和方法设置
样品制备 从各个大鼠腿部肌肉组织切片取样,以时间顺序增加进行尸检 及采样。采用氯仿/甲醇/水(1:3:1)提取经匀质的组织切片中 的代谢物,在冰块下孵育 1 小时。蛋白质和 DNA 进行离心沉 淀。去除上清液,贮藏在 -80 ℃ 条件下备用。
2 样品衍生化 移取 200 µL 提取液至 1.5 mL 硼硅玻璃小瓶中,加盖 9 mm 螺 丝盖。然后,将样品置于 Thermo Scientific™ Reacti-Vap™ 蒸 发器中,在 30 ℃ 条件下以低流速氮气流干燥 60 min。以下所 有衍生化步骤均采用 Thermo Scientific™ TriPlus™ RSH 自动 样品处理器进行。 加入 20 µL 20 mg/mL(w/v)甲氧基胺盐酸盐的吡啶溶液至 每个干燥小瓶中。将小瓶涡旋 10 秒并在 30 ℃ 条件下孵育 60 min。甲氧基化步骤完成以后,加入 30 µL MSTFA +1% TMCS(N-甲基-N-(三甲基硅基)三氟乙酰胺+1%三甲基氯 硅烷)的混合液,进一步涡旋 30 秒。将小瓶置于 45 ℃ 条件 下孵育 60 min 进行硅烷化。将样品冷却至室温以备进样。
基峰质量数 RT
最大强度
T0
T1
T2
T3
T0:T0
T1:T0
T2:T0
T3:T0
219.1100 10.71 507930367 1.0 2.7 3.4 4.5
1.000
0.008
0.000
0.003
232.1184 11.78 291673570 1.0 1.5 1.6 2.1
1.000
0.043
GC-MS 分析 所有实验均采用 Thermo Scientific™ Q Exactive™ GC 组合型 四极杆 Orbitrap 质谱仪进行。采用 TriPlus RSH 自动进样器 进样,采用 Thermo Scientific™ TRACE™ 1310 GC 色谱仪和 Thermo Scientific™ TraceGOLD™ TG-5SilMS 15 m × 0.25 mm I.D. × 0.25 µm 薄膜毛细管柱(P/N: 26096-1301)进行 色谱分离。仪器参数的其他详细信息见表 1 和表 2。
关键词
法医学;代谢组学;多变量统计分析;Q Exactive GC 系统
引言
代谢组学旨在表征和定量生物系统中的完整小分子代谢通路或 代谢物组。代谢物组包含小分子多元混合物(包括氨基酸、糖 和磷酸糖、生物胺和脂质)。非靶向代谢组学极具挑战性,因 为其要求定性和定量上百个不同种类化合物,而有关这些代谢 物的经验知识有限。因此,有必要使用一个既可以以非靶向的 方式灵敏检测特定分子,又可以提供精确质量数信息,用于确 认未知物并对其进行结构解析的检测系统。
图 6. 分解数据的 PLS-DA 模型。该模型为有监督多变量分析,将高维数据(例如,大量强度不同的代谢物)折叠为涵盖数据集中主要变化的几种主 成分。在本例中,X 轴是主成分 1,Y 轴是主成分 2。注意,本项研究对样品进行了适当聚类,每个群组聚集在一起,T0 已明显与其他群组分开。
0.06
● W*c [2]
T0 T1 T2 T3 30
20
10
t [2]
0
-10
-20
-30
-40 -80
-60
-40
-20
0
20
40
60
t [1]
R2ⅹ [1] = 0.385
R2ⅹ [2] = 0.127
E11ipse: Hote11ing's T2 (95%)
SIMCA 13.0-31/03/2015 11:47:15(UTC-1)
P 值(T 检验)
4.00E+008
2.00E+008
0.00E+000 16.92232 16.97232
17.02232
17.07232 17.12232 17.17232
图 3. 已解卷积峰簇被推断识别为酪氨酸。
17.22232
T0/T3
强度比
图 4. 发生显著性变化代谢物的火山图。每个代谢物以深蓝色菱形表示。 强度比(X 轴)是指 T0/T3 时代谢物的倍数变化,Y 轴显示代谢物的 P 值。因此,右上方和左上角的代谢物是倍数变化最大且最具有统计学意 义的代谢物。
1.000
0.015
0.001
0.026
156.1203 16.88 1169362271 1.0 3.6 3.9 5.1
1.000
0.000
0.000
0.007
5
使用 SIMCA 软件进行多变量统计分析。5将结论数据转化为更为直观的对数值,将 Y 类别设为各时间点,生成数据的偏最小二乘 判别分析(PLS-DA)模型。该软件生成分数图和载荷图,以显示沿着主成分每个时间点的聚类和分离(图 6)情况。从本次分析 可知,死亡后立即取样的样品(RAT_T0)聚集在一起,与已分解的大鼠样品(T1–T3)具有显著性差异。从 T1–T3 样品中可以观 察到分组聚类和持续分解。X 或 Y 轴上的位移表示某一代谢物对分数图中样品群组之间分离的作用大小。在本例中,X 轴将 T0 与 T1–3 分开,Y 轴将 T1、T2 和 T3 分开(图 6)。载荷图上的每个蓝点表示每个已检测到的含 EI 碎片离子簇的代谢物(图 7)。
本项研究展示了新型 Thermo Scientific™ Orbitrap™ GC-MS 完整非靶向代谢组学工作流程,检测了大鼠模型中的生物标记 物,以判定其死亡时间。死亡时间(PMI)推断是法医调查中 一项最关键也是最困难的任务,尤其当尸体温度与周围环境温 度到平衡后。由于当前用于确定 PMI 的方法不精确性且以 目视检查尸体为主。因此,建立一种实验方法,使用耐用生物 标志物推断死亡时间可辅助法医调查。
定量阶段 首先使用单变量统计方法分析结果。使用 Student's t 检验对比 每个时间点和时间零点。将每个代谢物的平均 T0 强度设为 1, 倍数变化显示为 1 的倍数值,以方便对比强度明显不同的代谢 物(图 4 和图 5)。已检测到 272 个代谢物的平均强度发生显 著性变化(P 值小于 0.05),包含已检测峰簇的定量矩阵示例 见表 3。
表 3. 已检测峰簇的定量矩阵。基峰(峰簇中强度最大的峰)、保留时间(RT)和检测到的最大强度见第 1–3 列。将 T0 的强度归一化为 1,其他 时间点的强度与 T0 强度相比并视情况以不同颜色显示,见第 4–7 列。第 8–11 列包含每项比值的 T 检验 P 值。
T 检验: T 检验: T 检验: T 检验:
0.04
0.02
W*c [2]
0.055
0.030
232.1184 12.41 941914122 1.0 5.9 8.5 11.0
1.000
0.013
0.001
0.002
156.0840 15.32 1163630714 1.0 2.1 1.9 2.0
1.000
0.019
0.008
0.021
174.1128 16.18 203636192 1.0 2.5 3.2 5.0
结果和结论
将八只大鼠尸体置于室温下四天。每日提取肌肉组织进行分 析,共检测 16 个样品中代谢物浓度的变化以分析尸体分解程 度。对样品进行随机分析以减小系统误差。代谢组学优化后的 分析流程见图 1。
表 1. GC 温度程序
TRACE 1310 GC 参数
进样量(µL)
1.0
衬管
单锥形(P/N 453A1345)
气相色谱质谱联用仪(GC-MS)基于其自身的优点经常用于 代谢组学分析,尤其是其色谱分辨率、重现性、峰容量和便于 使用的质谱库。GC为非靶向代谢组学中生物标志物的发现提 供了卓越的色谱分离能力,但是,由于目前缺乏高端质谱仪的 支持,从而限制了提供用于分析复杂样品(例如,哺乳动物的 肌肉组织)的动态范围、精确质量数和扫描速度。鉴于大部分 重要代谢物的极性特征,必须将其衍生化,以使其有效挥发, 从而确保得到良好的色谱分离。代谢组学分析尤其是临床代谢 组学要求高通量和先进的自动化技术。
AN 10457_C_GCMSMS_201508Y
基于 Orbitrap GC-MS 的非靶向代谢组学
Stefan Weidt,2 Bogusia Pesko,2 Cristian Cojocariu,1 Paul Silcock,1 Richard J. Burchmore,2 and Karl Burgess2 1Thermo Fisher Scienti c, Runcorn, UK 2Glasgow Polyomics, University of Glasgow, Glasgow, UK
数据处理
数据分析流程首先使用 MSConvert(ProteoWizard 套件1的一 部分)将原始数据文件转换为 MzXML 文件。使用 XCMS 包2 和 centWave 算法提取峰。已检测到的峰以 PeakML 格式输出, 然后使用 MzMatch.R 包对已检测到的峰进行后处理(过滤每 组重复进样的最低检测值、相对标准偏差和对 EI 碎片组进行分 组的相关性匹配)。3生成的文本文件输出使用 IDEOM 软件4 进 行单变量统计处理,使用 SIMCA™ 13.0.35 进行多变量统计处 理。使用 Thermo Scientific 解卷积软件进行峰簇识别和解析。