USP美国药典 233元素杂质-检查法
美国药典重金属检查法

231HEAVY METALSThis test is provided to demonstrate that the content of metallic impurities that are colored by sulfide ion, under the specified test conditions, does not exceed the Heavy metals limit specified in the individual monograph in percentage (by weight) of lead in the test substance, as determined by concomitant visual comparison (see Visual Comparison in the section Procedure underSpectrophotometry and Light-Scattering 851) with a control prepared from a Standard Lead Solution. [NOTE—Substances that typically will respond to this test are lead, mercury, bismuth, arsenic, antimony, tin, cadmium, silver, copper, and molybdenum.]Determine the amount of heavy metals by Method I, unless otherwise specified in the individual monograph. Method I is used for substances that yield clear, colorless preparations under the specified test conditions. Method II is used for substances that do not yield clear, colorless preparations under the test conditions specified for Method I, or for substances that, by virtue of their complex nature, interfere with the precipitation of metals by sulfide ion, or for fixed and volatile oils. Method III, a wet-digestion method, is used only in those cases where neither Method I nor Method II can be used.Special ReagentsLead Nitrate Stock Solution— Dissolve 159.8 mg of lead nitrate in 100 mL of water to which has been added 1 mL of nitric acid, then dilute with water to 1000 mL. Prepare and store this solution in glass containers free from soluble lead salts.Standard Lead Solution— On the day of use, dilute 10.0 mL of Lead Nitrate Stock Solution with water to 100.0 mL. Each mL of Standard Lead Solution contains the equivalent of 10 µg of lead. A comparison solution prepared on the basis of 100 µL of Standard Lead Solution per g of substance being tested contains the equivalent of 1 part of lead per million parts of substance being tested.METHOD IpH 3.5 Acetate Buffer— Dissolve 25.0 g of ammonium acetate in 25 mL of water, and add 38.0 mL of 6 N hydrochloric acid. Adjust, if necessary, with 6 N ammonium hydroxide or 6 N hydrochloric acid to a pH of 3.5, dilute with water to 100 mL, and mix.Standard Preparation— Into a 50-mL color-comparison tube pipet 2 mL of Standard Lead Solution (20 µg of Pb), and dilute with water to 25 mL. Using a pH meter or short-range pH indicator paper as external indicator, adjust with 1 N acetic acid or 6 N ammonium hydroxide to a pH between 3.0 and 4.0, dilute with water to 40 mL, and mix.Test Preparation— Into a 50-mL color-comparison tube place 25 mL of the solution prepared for the test as directed in the individual monograph; or, using the designated volume of acid where specified in the individual monograph, dissolve in and dilute with water to 25 mL the quantity, in g, of the substance to be tested, as calculated by the formula:2.0/(1000L),in which L is the Heavy metals limit, as a percentage. Using a pH meter or short-range pH indicator paper as external indicator, adjust with 1 N acetic acid or 6 N ammonium hydroxide to a pH between 3.0 and 4.0, dilute with water to 40 mL, and mix.Monitor Preparation— Into a third 50-mL color-comparison tube place 25 mL of a solution prepared as directed for Test Preparation, and add 2.0 mL of Standard Lead Solution. Using a pH meter or short-range pH indicator paper as external indicator, adjust with 1 N acetic acid or 6 N ammonium hydroxide to a pH between 3.0 and 4.0, dilute with water to 40 mL, and mix. Procedure— To each of the three tubes containing the Standard Preparation, the Test Preparation, and the Monitor Preparation, add 2 mL of pH 3.5 Acetate Buffer, then add 1.2 mL of thioacetamide–glycerin base TS, dilute with water to 50 mL, mix, allow to stand for 2 minutes, and view downward over a white surface *: the color of the solution from the Test Preparation is not darker than that of the solution from the Standard Preparation, and the color of the solution from the Monitor Preparation is equal to or darker than that of the solution from the Standard Preparation. [NOTE—If the color of the Monitor Preparation is lighter than that of the Standard Preparation, use Method II instead of Method I for the substance being tested.]METHOD IINOTE—This method does not recover mercury.pH 3.5 Acetate Buffer— Prepare as directed under Method I.Standard Preparation— Pipet 4 mL of the Standard Lead Solution into a suitable test tube, and add 10 mL of 6 N hydrochloric acid.Test Preparation— Use a quantity, in g, of the substance to be tested as calculated by the formula:4.0/(1000L),in which L is the Heavy metals limit, as a percentage. Transfer the weighed quantity of the substance to a suitable crucible, add sufficient sulfuric acid to wet the substance, and carefully ignite at a low temperature until thoroughly charred. (The crucible may be loosely covered with a suitable lid during the charring.) Add to the carbonized mass 2 mL of nitric acid and 5 drops of sulfuric acid, and heat cautiously until white fumes no longer are evolved. Ignite, preferably in a muffle furnace, at 500to 600, until the carbon is completely burned off (no longer than 2 hours). If carbon remains, allow the residue to cool, add a few drops of sulfuric acid, evaporate, and ignite again. Cool, add 5 mL of 6 N hydrochloric acid, cover, and digest on a steam bath for 10 minutes. Cool, and quantitatively transfer the solution to a test tube. Rinse the crucible with a second 5-mL portion of 6 N hydrochloric acid, and transfer the rinsing to the test tube.Monitor Preparation— Pipet 4 mL of the Standard Lead Solution into a crucible identical to that used for the Test Preparation and containing a quantity of the substance under test that is equal to 10% of the amount required for the Test Preparation. Evaporate on a steam bath to dryness. Ignite at the same time, in the same muffle furnace, and under the same conditions used for the Test Preparation. Cool, add 5 mL of 6 N hydrochloric acid, cover, and digest on a steam bath for 10 minutes. Cool, and quantitatively transfer to a test tube. Rinse the crucible with a second 5-mL portion of 6 N hydrochloric acid, and transfer the rinsing to the test tube.Procedure— Adjust the solution in each of the tubes containing the Standard Preparation, the Test Preparation, and the Monitor Preparation with ammonium hydroxide, added cautiously and dropwise, to a pH of 9. Cool, and adjust with glacial acetic acid, added dropwise, to a pH of 8, then add 0.5 mL in excess. Using a pH meter or short-range pH indicator paper as external indicator, check the pH, and adjust, if necessary, with 1 N acetic acid or 6 N ammonium hydroxide to a pH between 3.0 and 4.0. Filter, if necessary, washing the filter with a few mL of water, into a 50-mL color-comparison tube, and then dilute with water to 40 mL. Add 2 mL of pH 3.5 Acetate Buffer, then add 1.2 mL of thioacetamide–glycerin base TS, dilute with water to 50 mL, mix, allow to stand for 2 minutes, and view downward over a white surface*: the color of the solution from the Test Preparation is not darker than that of the solution from the Standard Preparation, and the color of the solution from the Monitor Preparation is equal to or darker than that of the solution from the Standard Preparation. [NOTE—If the color of the solution from the Monitor Preparation is lighter than that of the solution from the Standard Preparation, proceed as directed for Method III for the substance being tested.]METHOD IIIpH 3.5 Acetate Buffer— Prepare as directed under Method I.Standard Preparation— Transfer a mixture of 8 mL of sulfuric acid and 10 mL of nitric acid to a clean, dry, 100-mL Kjeldahl flask, and add a further volume of nitric acid equal to the incremental volume of nitric acid added to the Test Preparation. Heat the solution to the production of dense, white fumes; cool; cautiously add 10 mL of water; and, if hydrogen peroxide was used in treating the Test Preparation, add a volume of 30 percent hydrogen peroxide equal to that used for the substance being tested. Boil gently to the production of dense, white fumes. Again cool, cautiously add 5 mL of water, mix, and boil gently to the production of dense, white fumes and to a volume of 2 to 3 mL. Cool, dilute cautiously with a few mL of water, add 2.0 mL of Standard Lead Solution (20 µg of Pb), and mix. Transfer to a 50-mL color-comparison tube, rinse the flask with water, adding the rinsing to the tube until the volume is 25 mL, and mix.Test Preparation— Unless otherwise indicated in the individual monograph, use a quantity, in g, of the substance to be tested as calculated by the formula:2.0/(1000L),in which L is the Heavy metals limit, as a percentage.If the substance is a solid— Transfer the weighed quantity of the test substance to a clean, dry, 100-mL Kjeldahl flask. [NOTE—A 300-mL flask may be used if the reaction foams excessively.] Clamp the flask at an angle of 45, and add a sufficient quantity of a mixture of 8 mL of sulfuric acid and 10 mL of nitric acid to moisten the substance thoroughly. Warm gently until the reaction commences, allow the reaction to subside, and add portions of the same acid mixture, heating after each addition, until a total of 18 mL of the acid mixture has been added. Increase the amount of heat, and boil gently until the solution darkens. Cool, add 2 mL of nitric acid, and heat again until the solution darkens. Continue the heating, followed by addition of nitric acid until no further darkening occurs, then heat strongly to the production of dense, white fumes. Cool, cautiously add 5 mL of water, boil gently to the production of dense, white fumes, and continue heating until the volume is reduced to a few mL. Cool, cautiously add 5 mL of water, and examine the color of the solution. If the color is yellow, cautiously add 1 mL of 30 percent hydrogen peroxide, and again evaporate to the production of dense, white fumes and a volume of 2 to 3 mL. If the solution is still yellow, repeat the addition of 5 mL of water and the peroxide treatment. Cool, dilute cautiously witha few mL of water, and rinse into a 50-mL color-comparison tube, taking care that the combined volume does not exceed 25 mL.If the substance is a liquid— Transfer the weighed quantity of the test substance to a clean, dry, 100-mL Kjeldahl flask. [NOTE—A 300-mL flask may be used if the reaction foams excessively.] Clamp the flask at an angle of 45, and cautiously add a few mL of a mixture of 8 mL of sulfuric acid and 10 mL of nitric acid. Warm gently until the reaction commences, allow the reaction to subside, and proceed as directed for If the substance is a solid,beginning with ―add portions of the same acid mixture.‖Monitor Preparation— Proceed with the digestion, using the same amount of sample and the same procedure as directed in the subsection If the substance is a solid in the section Test Preparation, until the step ―Cool, dilute cautiously with a few mL of water.‖ Add 2.0 mL of Lead Standard Solution (20 µg of lead), and mix. Transfer to a 50-mL color comparison tube, rinse the flask with water, adding the rinsing to the tube until the volume is 25 mL, and mix. Procedure— Treat the Test Preparation, the Standard Preparation, and the Monitor Preparation as follows. Using a pH meter or short-range pH indicator paper as external indicator, adjust the solution to a pH between 3.0 and 4.0 with ammonium hydroxide (a dilute ammonia solution may be used, if desired, as the specified range is approached), dilute with water to 40 mL, and mix.To each tube add 2 mL of pH 3.5 Acetate Buffer, then add 1.2 mL of thioacetamide–glycerin base TS, dilute with water to 50 mL, mix, allow to stand for 2 minutes, and view downward over a white surface*: the color of the Test Preparation is not darker than that of the Standard Preparation, and the color of the Monitor Preparation is equal to or darker than that of the Standard Preparation.。
232、233 草案验证在测定原料药元素中的应用

摘要美国药典 (USP) 组织正在开发有关药品和原料药中元素杂质测定的新通则。
USP<232> 规定了分析物的限量,而 USP<233> 则规定了样品制备选项(包括密闭容器微波消解)并推荐使用现代仪器,例如多元素 ICP-MS 和 ICP-OES 技术。
根据 USP<233> 的规定,分析仪器的检定须基于性能测试,包括需要论证仪器的准确性、重现性以及能够可靠鉴定分析物。
在本文中,我们列出的数据表明依照 USP<232>/<233> 成功验证了 Agilent 7700x ICP-MS 检测明胶胶囊样品中元素杂质的应用。
参照美国药典通则 <232>/<233> 草案验证 Agilent 7700x ICP-MS 在测定原料药元素杂质中的应用应用简报作者Samina Hussain Exova 美国Amir Liba and Ed McCurdy 安捷伦科技公司 美国制药所有规定的元素都通过了 USP<233> 的可接受标准,包括那些可能遭受基质干扰(比如 Cl 叠加在 V、Cr 和 As 上)的元素。
在 USP<232> 所要求的元素中,有些在氯化物基质中更易溶解或在化学上更加稳定,尤其是低浓度汞和铂族元素 (PGE),因此可在所有水溶液和酸消化样品中加入低百分比浓度的 HCl 使它们稳定。
由于通过氦反应池气体(7700 的标准操作模式)能可靠地消除基于 Cl 的多原子干扰,因此,在常规操作中加入 HCl 来稳定样品对于 ICP-MS 方法来说已不再是问题。
引言美国药典 (USP) 组织目前正在开发药物中无机(元素性)杂质的新监测方法。
新通则 USP<232>(限量)和 <233> (规程)将在 2013 年实施。
USP<232> 为覆盖面更广的无机(元素)杂质(As、Cd、Hg、Pb、V、Cr、Ni、Mo、Mn、Cu、Pt、Pd、Ru、Rh、Os 和 Ir)规定了新的、更低的允许日接触量 (PDE) [1]。
美国药典重金属检测方法-中文

铑
10
40
0.0001
钌
10
40
0.0002
铬
25
100
0.002
钼
25
100
0.0002
镍
25
100
0.002
钒
25
100
0.005
铜
250
1000
0.002
镁
250
1000
0.001
样品制备
范围广泛的各种样品都可以用 USP<232>/<233> 进行分 析,所以提供适合所有样品类型的详细样品处理方法并不现 实。有些药物样品可以直接分析(不用溶解),而其他样品 可以用水性溶剂(如水或稀酸)或适当的有机溶剂(如 2-丁 氧乙醇 : 水(25 : 75)[3],DMSO 或 DGME)简单稀释或 溶解进行制备。用水性或有机溶剂进行简单稀释或溶解的 方法必须考虑样品的化学稳定性,并且对于有机溶剂溶解, 还要考虑样品中组分化合物的不同挥发性。对许多 API 来 说,用有机溶剂稀释是首选方法,这种情况下有必要采取有 助于稳定分析物的方法,以避免因较高或较低挥发性(与校 正标准品相比)成分的存在而造成的回收率波动 [7]。
USP<232> 包括一个涉及元素形态的章节,指出 As 和 Hg 的某些形态值得关注,因为其毒性比其它形态要大得 多。As 的 PDE 是指无机 As,如果总 As 浓度超出限度, 必须用一种能够对不同 As 形态进行分离和定量的方法对样 品进行重新分析。这样做的原因是无机 As 比常见的有机形 式(如,砷甜菜碱)毒性大得多,因此形态分析必须能够分 离其不同化学形态,确定无机 As(亚砷酸盐(三价 As)和 砷酸盐(四价 As))的总量低于限量。同样,Hg 限量也是 指无机 Hg(Hg2+),虽然甲基汞(MeHg)是毒性更大的 形态,但通常认为药物中不可能存在 MeHg。但如果样品来 自于可能含有相当量甲基汞的原料(如,鱼组织),也必须 对其进行特别的分离和测定。
元素杂质方法验证

元素杂质方法验证
元素杂质是指不同于主要成分的微量元素或化合物。
为了保证药品的质量和安全性,需要对药品中的元素杂质进行验证。
以下是几种常用的元素杂质方法验证:
1. 原子荧光光谱法(AA):该方法是根据不同元素的原子吸收特性,利用吸收光谱测定其中某些元素的含量。
AA法具有灵敏度高、分辨率好、选择性强等优点,是常用的元素杂质分析方法之一。
2. 感应耦合等离子体质谱法(ICP-MS):该方法可以同时测定元素杂质的多个元素,具有高精度和高分析速度等优点。
3. 原子荧光光度法(AED):使用激发光源使样品中的元素发生激发态,然后测量其发射光谱,从而确定其中的元素种类和含量。
该方法具有很高的检测灵敏度和多元素分析的能力。
4. 燃烧离子色谱法(CIC):CIC法主要用于测定药品中的卤素含量,该方法具有独特的分离能力和极高的灵敏度。
5. 紫外-可见分光光度法(UV-Vis):该方法主要用于有色元素的检测,如铁、铜、镍等金属离子。
通过测量样品的吸收光谱,可以确定其中的元素含量。
usp232_233原文 - 翻译

232元素杂质—限度介绍本通则明确了药品中各元素杂质的限度。
元素杂质包括催化剂、环境污染物,可能存在于原料药、赋形剂、制剂中。
这些杂质或自然产生,或有意添加,或由于不注意而引入(例如,与处理设备相互作用)。
当知道元素杂质存在,或有意添加,或有引入的可能性,应当保证这些杂质符合限度要求。
可以采用基于风险的控制策略来确保产品满足限度标准。
由于砷、镉、铅和汞普遍存在的特性,风险控制策略至少应考虑这四种元素。
不管采用什么分析方法,所有药品均应满足元素杂质限度标准。
本章提出的限度标准不适用于赋形剂与原料药,除非本章或各论中明确说明。
然而赋形剂与原料药中元素杂质水平必须报告。
本章提出的限度标准同样不适用于兽用产品和常规疫苗。
饮食补充剂和它们的成份的相关规定见于《饮食补充剂中的元素杂质》2232 .1形态分析对于元素氧化态、有机络合态、化合态的测定,称为形态分析。
每种元素可能存在不同的氧化态或络合态。
然而,砷和汞应特别关注,因为它们的无机态和络合有机态具有不同的毒性。
砷的限度标准是基于无机态(毒性最大)。
假定样品中所有砷都是无机态,可用总砷测定法检测。
当总砷法结果超过限度标准,应当使用能够对不同形态砷定量的分析方法,以确定无机态砷是否满足法定要求。
汞的限度标准是基于无机(2+)氧化态。
甲基汞(毒性最强),但对于药品,通常不是问题。
这样,汞限度标准的确定是基于汞最常见的无机形态。
对于可能含有甲基汞的产品(例如,从鱼中得到的物质),相应的汞限度标准将在各论中提及。
接触途径元素杂质的毒性跟接触程度(生物利用度)有关。
对于每一种元素杂质接触程度取决于给药途径:口服、肠外注射、吸入。
这些限度确定是基于慢性接触。
为建立标准需要,另两种给药途径,黏膜和局部接触可认为跟口服相同,而表1中的PDE值也适用于这些产品[注意—药品的给药途径在制剂通则中介绍制剂通则1151. ]Change to read:药品表1的第二至第四栏给出的限度值是一些元素杂质的基本日剂量PDE值(病人按指定给药途径服用)。
食品药品原料中元素杂质的法规要求与控制方法

原料药中元素杂质的法规要求及控制方法张再奇元素杂质又称重金属,重金属原义指比重大于5的金属,元素杂质包括可能存在于原料、辅料或制剂中,来源于合成中催化剂残留、药品生产制备过程中引入或辅料中存在的、生产设备引入、或容器密闭系统引入。
某些元素杂质不仅对药品的稳定性、保质期产生不利影响,还可能因为潜在的毒性引发药物副反应。
因此欧盟、美国对杂质的控制越来越严格,对此项不断修订,中国在加入ICH后对此项检测应该也会向国际靠拢,因此了解法规对元素杂质的要求、建立有效的检测方法变得尤为重要。
一、各国法规变更史(1)EMA、EP关于元素杂质的修订EP最新版为9.0版,其中保留了2.4.8金属测试方法A-H;2.4.20章节金属催化剂和金属试剂残留检测;5.20金属催化剂或金属试剂残留。
但在9.3增补版(2018年1月1日实施)中5.20项下规定,元素杂质限度遵循ICH要求。
EMA对元素杂质的修订如下表1。
(2)ICH对元素杂质的修订历程ICH于2009年10月批准了Q3D,经多方讨论后,修订版本的Q3D step4于2014年12月16日生效,其中列出了24种元素杂质的三种给药途径的PDE 值,确定实施日期为:新上市许可为2016年6月生效,已上市品种为2017年12月生效。
(3)USP对元素杂质的修订历程FDA规定在2018年1月1日之后,针对USP药典品种,提交新的NDA、ANDA 应该符合USP<232>、<233>。
针对非USP药典品种,申请人提交新的NDA、ANDA时,应该遵循Q3D。
美国对元素杂质的规定与ICH规定在不同时期,内容不一致,但从2017年12月之后,USP对元素种类和限量均与ICH保持一致。
修订历程详见下表2。
(4)中国药典对重金属检测的修订中国药典对重金属检测的修订主要体现在表3中,名称仍然为重金属,方法仍采用比色法,2017年中国成为了ICH成员国,未来中国的药政监管将遵循ICH指南规定,元素杂质与国际接轨也是大势所趋。
重金属检查法(USP和EP)
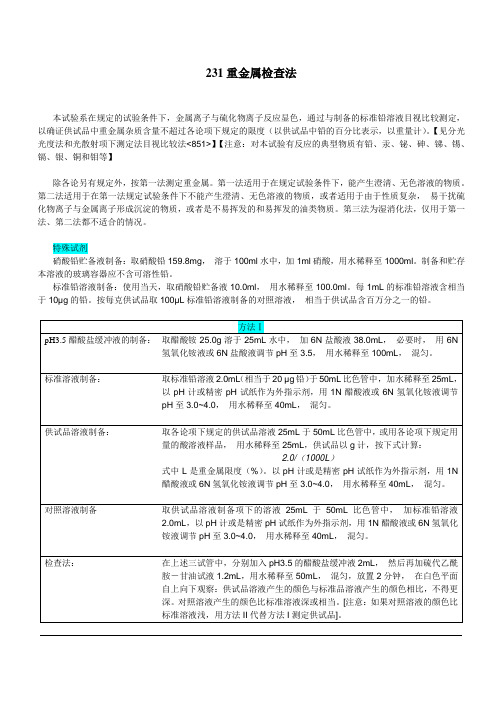
231重金属检查法本试验系在规定的试验条件下,金属离子与硫化物离子反应显色,通过与制备的标准铅溶液目视比较测定,以确证供试品中重金属杂质含量不超过各论项下规定的限度(以供试品中铅的百分比表示,以重量计)。
【见分光光度法和光散射项下测定法目视比较法<851>】【注意:对本试验有反应的典型物质有铅、汞、铋、砷、锑、锡、镉、银、铜和钼等】除各论另有规定外,按第一法测定重金属。
第一法适用于在规定试验条件下,能产生澄清、无色溶液的物质。
第二法适用于在第一法规定试验条件下不能产生澄清、无色溶液的物质,或者适用于由于性质复杂,易干扰硫化物离子与金属离子形成沉淀的物质,或者是不易挥发的和易挥发的油类物质。
第三法为湿消化法,仅用于第一法、第二法都不适合的情况。
特殊试剂硝酸铅贮备液制备:取硝酸铅159.8mg,溶于100ml水中,加1ml硝酸,用水稀释至1000ml。
制备和贮存本溶液的玻璃容器应不含可溶性铅。
标准铅溶液制备:使用当天,取硝酸铅贮备液10.0ml,用水稀释至100.0ml。
每1mL的标准铅溶液含相当于10µg的铅。
按每克供试品取100µL标准铅溶液制备的对照溶液,相当于供试品含百万分之一的铅。
在上述二试管中,分别加入pH3.5的醋酸盐缓冲液2mL,然后再加硫代乙酰胺—甘油试液1.2mL,用水稀释至50mL,混匀,放置2分钟,在白色平面自上向下观察:供试品溶液产生的颜色与标准品溶液产生的颜色相比,不得更深。
EP 版的重金属分析方法重金属方法A供试溶液:12ml待测水溶液,2ml pH为3.5的缓冲溶液,混合后加1.2ml的硫代乙酰胺试液,立即混合。
对照溶液:10ml的标准铅溶液(1ppm or 2ppm Pb),2ml pH为3.5的缓冲溶液,2ml的待测液,混合后加1.2ml的硫代乙酰胺试液,立即混合。
空白溶液:10ml的水,2ml pH为3.5的缓冲溶液,2ml的测试溶液。
最新usp232-233原文---翻译

232元素杂质—限度123介绍4本通则明确了药品中各元素杂质的限度。
元素杂质包括催化剂、环境污染物,5可能存在于原料药、赋形剂、制剂中。
这些杂质或自然产生,或有意添加,或6由于不注意而引入(例如,与处理设备相互作用)。
当知道元素杂质存在,或7有意添加,或有引入的可能性,应当保证这些杂质符合限度要求。
可以采用基8于风险的控制策略来确保产品满足限度标准。
由于砷、镉、铅和汞普遍存在的9特性,风险控制策略至少应考虑这四种元素。
不管采用什么分析方法,所有药10品均应满足元素杂质限度标准。
11本章提出的限度标准不适用于赋形剂与原料药,除非本章或各论中明确说12明。
然而赋形剂与原料药中元素杂质水平必须报告。
13本章提出的限度标准同样不适用于兽用产品和常规疫苗。
饮食补充剂和它们14的成份的相关规定见于《饮食补充剂中的元素杂质》2232 .11516形态分析17对于元素氧化态、有机络合态、化合态的测定,称为形态分析。
每种元素可能存在不同的氧化态或络合态。
然而,砷和汞应特别关注,因为它们的无机态1819和络合有机态具有不同的毒性。
20砷的限度标准是基于无机态(毒性最大)。
假定样品中所有砷都是无机态,可用总砷测定法检测。
当总砷法结果超过限度标准,应当使用能够对不同形态2122砷定量的分析方法,以确定无机态砷是否满足法定要求。
23汞的限度标准是基于无机(2+)氧化态。
甲基汞(毒性最强),但对于药品,24通常不是问题。
这样,汞限度标准的确定是基于汞最常见的无机形态。
对于可25能含有甲基汞的产品(例如,从鱼中得到的物质),相应的汞限度标准将在各26论中提及。
27282930接触途径31元素杂质的毒性跟接触程度(生物利用度)有关。
对于每一种元素杂质接触程度取决于给药途径:口服、肠外注射、吸入。
这些限度确定是基于慢性接触。
3233为建立标准需要,另两种给药途径,黏膜和局部接触可认为跟口服相同,而表1中的PDE值也适用于这些产品[注意—药品的给药途径在制剂通则中介绍制剂通34则1151. ]35Change to read:363738药品39表1的第二至第四栏给出的限度值是一些元素杂质的基本日剂量PDE值(病40人按指定给药途径服用)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
á233ñ ELEMENTAL IMPURITIES—PROCEDURESINTRODUCTIONThis chapter describes two analytical procedures (Procedures 1 and 2) for the evaluation of the levels of the elemental impuri-ties. The chapter also describes criteria for acceptable alternative procedures. By means of validation studies, analysts will confirm that the analytical procedures described herein are suitable for use on specified material.Use of Alternative ProceduresThe chapter also describes criteria for acceptable alternative procedures. Alternative procedures that meet the validation re-quirements herein may be used in accordance with General Notices and Requirements 6.30, Alternative and Harmonized Meth-ods and Procedures . Information on the Requirements for Alternate Procedure Validation is provided later in this chapter.SpeciationThe determination of the oxidation state, organic complex, or combination is termed speciation . Analytical procedures for spe-ciation are not included in this chapter, but examples may be found elsewhere in USP–NF and in the literature.PROCEDURES• C OMPENDIAL P ROCEDURES 1 AND 2System standardization and suitability evaluation using applicable reference materials should be performed on the day of analysis.Procedure and detection technique:Procedure 1 can be used for elemental impurities generally amenable to detection byinductively coupled plasma–atomic (optical) emission spectroscopy (ICP–AES or ICP–OES). Procedure 2 can be used for ele-mental impurities generally amenable to detection by ICP–MS. Before initial use, the analyst should verify that the proce-dure is appropriate for the instrument and sample used (procedural verification) by meeting the alternative procedure vali-dation requirements below.Sample preparation:Forms of sample preparation include Neat , Direct aqueous solution , Direct organic solution , and Indi-rect solution . The selection of the appropriate sample preparation depends on the material under test and is the responsibil-ity of the analyst. When a sample preparation is not indicated in the monograph, an analyst may use any of the followingappropriately validated preparation procedures. In cases where spiking of a material under test is necessary to provide an acceptable signal intensity, the blank should be spiked with the same Target elements , and where possible, using the same spiking solution. Standard solutions may contain multiple Target elements . [N OTE —All liquid samples should be weighed.]Neat:Used for liquids or alternative procedures that allow the examination of unsolvated samples.Direct aqueous solution:Used when the sample is soluble in an aqueous solvent.Direct organic solution:Used where the sample is soluble in an organic solvent.Indirect solution:Used when a material is not directly soluble in aqueous or organic solvents. Total metal extraction is the preferred sample preparation approach to obtain an Indirect solution . Digest the sample using the Closed vessel diges-tion procedure provided below or one similar to it. The sample preparation scheme should yield sufficient sample to allow quantification of each element at the limit specified in the corresponding monograph or chapter.Closed vessel digestion:This sample preparation procedure is designed for samples that must be digested in a Concen-trated acid using a closed vessel digestion apparatus. Closed vessel digestion minimizes the loss of volatile impurities. The choice of a Concentrated acid depends on the sample matrix. The use of any of the Concentrated acids may be appropri-ate, but each introduces inherent safety risks. Therefore, appropriate safety precautions should be used at all times.[N OTE —Weights and volumes provided may be adjusted to meet the requirements of the digestion apparatus used.]An example procedure that has been shown to have broad applicability is the following. Dehydrate and predigest 0.5 g of primary sample in 5 mL of freshly prepared Concentrated acid . Allow to sit loosely covered for 30 min in a fume hood.Add an additional 10 mL of Concentrated acid , and digest, using a closed vessel technique, until digestion or extraction is complete. Repeat, if necessary, by adding an additional 5 mL of Concentrated acid . [N OTE —Where closed vessel digestion is necessary, follow the manufacturer’s recommended procedures to ensure safe use.]Alternatively, leachate extraction may be appropriate with justification following scientifically validated metal disposition studies, which may include animal studies, speciation, or other means of studying disposition of the specific metal in the drug product.Reagents:All reagents used for the preparation of sample and standard solutions should be free of elemental impurities,in accordance with Plasma Spectrochemistry á730ñ.• P ROCEDURE 1: ICP–OESStandardization solution 1: 1.5J of the Target element(s) in a Matched matrixStandardization solution 2:0.5J of the Target element(s) in a Matched matrixSample stock solution:Proceed as directed in Sample preparation above. Allow the sample to cool, if necessary. For mer-cury determination, add an appropriate stabilizer.Sample solution:Dilute the Sample stock solution with an appropriate solvent to obtain a final concentration of the Target elements at NMT 1.5J .Blank:Matched matrix298 á233ñ Elemental Impurities—Procedures / Chemical Tests USP 40Elemental spectrometric system(See Plasma Spectrochemistry á730ñ.)Mode:ICPDetector:Optical detection systemRinse:Diluent usedStandardization:Standardization solution 1, Standardization solution 2, and BlankSystem suitabilitySample:Standardization solution 1Suitability requirementsDrift:Compare results obtained from Standardization solution 1 before and after the analysis of the Sample solution.Suitability criteria:NMT 20% for each Target element. [N OTE—If samples are high in mineral content, rinse system well before introducing the Sample in order to minimize carryover.]Analysis:Analyze according to the manufacturer's suggestions for program and wavelength. Calculate and report results on the basis of the original sample size. [N OTE—Appropriate measures must be taken to correct for matrix-induced inter-ferences (e.g., wavelength overlaps).]• P ROCEDURE2: ICP–MSStandardization solution 1: 1.5J of the Target element(s) in a Matched matrixStandardization solution 2:0.5J of the Target element(s) in a Matched matrixSample stock solution:Proceed as directed for Sample preparation above. Allow the sample to cool, if necessary. For mercury determination, add an appropriate stabilizer.Sample solution:Dilute the Sample stock solution with an appropriate solvent to obtain a final concentration of the Target elements at NMT 1.5J.Blank:Matched matrixElemental spectrometric system(See Plasma Spectrochemistry á730ñ.)Mode:ICP. [N OTE—An instrument with a cooled spray chamber is recommended. (A collision cell or reaction cell may also be beneficial.)]Detector:Mass spectrometerRinse:Diluent usedStandardization:Standardization solution 1, Standardization solution 2, and BlankSystem suitabilitySample:Standardization solution 1Suitability requirementsDrift:Compare results obtained from Standardization solution 1 before and after the analysis of the Sample solution.Suitability criteria:Drift NMT 20% for each Target element. [N OTE—If samples are high in mineral content, rinse sys-tem well before introducing the Sample in order to minimize carryover.]Analysis:Analyze according to the manufacturer's suggestions for program and m/z. Calculate and report results based on the original sample size. [N OTE—Appropriate measures must be taken to correct for matrix-induced interferences (e.g., argon chloride interference with arsenic determinations).]REQUIREMENTS FOR ALTERNATE PROCEDURE VALIDATIONIf the specified compendial procedures do not meet the needs of a specific application, an alternative procedure may be devel-oped (see General Notices and Requirements 6.30, Alternative and Harmonized Methods and Procedures). Alternative proce-dures must be validated and shown to be acceptable, in accordance with the validation requirements for alternative proce-dures as described below. The level of validation necessary to ensure that an alternative procedure is acceptable depends on whether a limit test or a quantitative determination is specified in the monograph. The requirements for the validation of an elemental impurities procedure for each type of determination are described below. Any alternative procedure that has been validated and meets the acceptance criteria that follow is considered to be suitable for use.LIMIT PROCEDURESThe following section defines the validation parameters for the acceptability of alternative limit procedures. Meeting these re-quirements must be demonstrated experimentally using an appropriate system suitability procedure and reference material. The suitability of the method must be determined by conducting studies with the material or mixture under test supplemen-ted with known concentrations of each Target element of interest at the appropriate acceptance limit concentration. The ma-terial or mixture under test must be spiked before any sample preparation steps are performed.• D ETECTABILITYStandard solution: A preparation of reference materials for the Target element(s) at the Target concentrationSpiked sample solution 1:Prepare a solution of sample under test, spiked with appropriate reference materials for the Target elements at the Target concentration, solubilized or digested as described in Sample preparation.Spiked sample solution 2:Prepare a solution of the sample under test, spiked with appropriate reference materials at 80% of the Target concentration for the Target elements, solubilized or digested as described in Sample preparation.Unspiked sample solution: A sample of material under test, solubilized or digested in the same manner as the SamplesolutionsUSP 40Chemical Tests / á233ñ Elemental Impurities—Procedures 299Acceptance criteriaNon-instrumental procedures:Spiked sample solution 1 provides a signal or intensity equivalent to or greater than that of the Standard solution . Spiked sample solution 2 must provide a signal or intensity less than that of Spiked sample solu-tion 1. [N OTE —The signal from each Spiked sample solution is NLT the Unspiked sample solution determination.]Instrumental procedures:The average value of the three replicate measurements of Spiked sample solution 1 is within ±15% of the average value obtained for the replicate measurements of the Standard solution . The average value of the replicate measurements of Spiked sample solution 2 must provide a signal intensity or value less than that of the Standard solution . [N OTE —Correct the values obtained for each of the spiked solutions using the Unspiked sample solution .]• P RECISION FOR I NSTRUMENTAL M ETHODS (R EPEATABILITY )[N OTE —Non-instrumental precision is demonstrated by meeting the Detectability requirement above.]Sample solutions:Six independent samples of the material under test, spiked with appropriate reference materials for the Target elements at the Target concentrationAcceptance criteriaRelative standard deviation:NMT 20% for each Target element• S PECIFICITYThe procedure must be able to unequivocally assess (see Validation of Compendial Procedures á1225ñ) each Target element in the presence of components that may be expected to be present, including other Target elements , and matrix compo-nents.QUANTITATIVE PROCEDURESThe following section defines the validation parameters for the acceptability of alternative quantitative procedures. Meetingthese requirements must be demonstrated experimentally, using an appropriate system suitability procedure and referencematerials. Meeting these requirements demonstrates that the procedure is equivalent to the compendial procedure for thepurpose of quantifying the Target elements .• A CCURACYStandard solutions:Prepare solutions containing the Target elements at concentrations ranging from 50% to 150% of J ,using appropriate reference materials.Test samples:Prepare samples of the material under test spiked with appropriate reference materials before any sample preparation steps (digestion or solubilization) at concentrations ranging from 50% to 150% of J for each Target element .Acceptance criteriaSpike recovery:70%–150% for the mean of three replicate preparations at each concentration• P RECISIONRepeatabilityTest samples:Six independent samples of material under test (taken from the same lot) spiked with appropriate refer-ence materials for the Target element(s) at the indicated levelAcceptance criteriaRelative standard deviation:NMT 20% (N = 6) for each Target elementIntermediate precision (ruggedness)Perform the Repeatability analysis again either on a different day, with a different instrumentation, with a different analyst,or a combination thereof. Combine the results of this analysis with the Repeatability analysis so the total number of anal-yses is 12.Acceptance criteriaRelative standard deviation:NMT 25% (N = 12) for each Target element• S PECIFICITYThe procedure must be able to unequivocally assess (see á1225ñ) each Target element in the presence of components that may be expected to be present, including other Target elements , and matrix components.• L IMIT OF Q UANTITATION , R ANGE , AND L INEARITYDemonstrated by meeting the Accuracy requirement.GLOSSARYConcentrated acid:Concentrated ultra-pure nitric, sulfuric, hydrochloric, or hydrofluoric acids or Aqua regiaAqua regia:Aqua regia is a mixture of concentrated hydrochloric and nitric acids, typically at ratios of 3:1 or 4:1, respective-ly.Matched matrix:Solutions having the same solvent composition as the Sample solution . In the case of an aqueous solution,Matched matrix would indicate that the same acids, acid concentrations, and mercury stabilizer are used in both prepara-tions.Target elements:Elements with the potential of being present in the material under test. Include arsenic (As), cadmium (Cd), lead (Pb), and mercury (Hg) in the target element evaluation when testing is done to demonstrate compliance. Target elements should also include any elements that may be added through material processing or storage.Target limit or Target concentration:The acceptance value for the elemental impurity being evaluated. Exceeding the Tar-get limit indicates that a material under test exceeds the acceptable value. The determination of compliance is addressed in other chapters. [N OTE —When applying this chapter to Elemental Impurities—Limits á232ñ andElemental Contaminants in Diet-300 á233ñ Elemental Impurities—Procedures / Chemical Tests USP 40ary Supplements á2232ñ, Target limits can be approximated by dividing the Daily Dose PDEs by the maximum daily dose for the Drug Product Analysis Option in á232ñ or the Daily Serving PDE divided by the maximum daily serving size in á2232ñ.]J:The concentration (w/w) of the element(s) of interest at the Target limit, appropriately diluted to the working range of the instrument. For example, if the target elements are lead and arsenic for an analysis of an oral solid drug product with a daily dose of 10g/day using inductively coupled plasma–mass spectrometry (ICP–MS), the target limit for these elements would be 0.5 m g/g and 1.5 m g/g (see Table 2 in á232ñ). However, in this case, the linear dynamic range of the ICP–MS is known to extend from 0.01 ng/mL to 0.1 m g/mL for these elements. Therefore, a dilution factor of at least 1:100 is required to ensure that the analysis occurs in the linear dynamic range of the instrument. J would thus equal 5 ng and 15 ng/mL for lead and arsenic, respectively, when the dilution factor is added.Appropriate reference materials:Where Appropriate reference materials are specified in the chapter, certified reference ma-terials (CRM) from a national metrology institute (NMI), or reference materials that are traceable to the CRM of an NMI should be used. An example of an NMI in the United States is the National Institute of Standards and Technology.á241ñ IRONThis limit test is provided to demonstrate that the content of iron, in either the ferric or the ferrous form, does not exceed the limit for iron specified in the individual monograph. The determination is made by concomitant visual comparison with a control prepared from a standard iron solution.SPECIAL REAGENTSStandard Iron SolutionDissolve 863.4 mg of ferric ammonium sulfate [FeNH4(SO4)2·12H2O] in water, add 10 mL of 2N sulfuric acid, and dilute with water to 100.0 mL. Pipet 10 mL of this solution into a 1000-mL volumetric flask, add 10 mL of 2N sulfuric acid, dilute with water to volume, and mix. This solution contains the equivalent of 0.01 mg (10 m g) of iron per mL.Ammonium Thiocyanate SolutionDissolve 30g of ammonium thiocyanate in water to make 100 mL.STANDARD PREPARATIONInto a 50-mL color-comparison tube pipet 1 mL of Standard Iron Solution (10 m g of Fe), dilute with water to 45 mL, add 2 mL of hydrochloric acid, and mix.TEST PREPARATIONInto a 50-mL color comparison tube place the solution prepared for the test as directed in the individual monograph and if necessary dilute with water to 45 mL; or, dissolve in water, and dilute with water to 45 mL the quantity, in g, of the substance to be tested, as calculated by the formula:1.0/(1000L)in which L is the Iron limit in percentage. Add 2 mL of hydrochloric acid, and mix.PROCEDURETo each of the tubes containing the Standard Preparation and the Test Preparation add 50 mg of ammonium peroxydisulfate crystals and 3 mL of Ammonium Thiocyanate Solution, and mix: the color of the solution from the Test Preparation is not darker than that of the solution from theStandard Preparation.USP 40Chemical Tests / á241ñ Iron 301。