与注射剂再评价相关的包装完整性评估
中药注射剂再评价指导原则

中药注射剂再评价指导原则中药注射剂再评价指导原则引言:中药注射剂作为中药领域的重要代表之一,在临床应用中发挥着重要的作用。
然而,过去对于中药注射剂再评价指导原则的相关研究相对较少,导致临床上对于其应用的规范性和安全性存在一定的争议。
为了推动中药注射剂再评价工作的顺利进行,本文将围绕中药注射剂再评价的概念、目的、方法和意义展开深入探讨。
1. 中药注射剂再评价的概念中药注射剂再评价是指对已上市中药注射剂进行全面系统的再评估和再评价的工作。
它对于提高中药注射剂的质量和疗效,以及保障患者用药安全起到至关重要的作用。
2. 中药注射剂再评价的目的中药注射剂再评价的目的在于继续深入了解其药理学、药代动力学、药效学等方面的特点,全面掌握其临床应用的风险和效益,并为临床合理应用中药注射剂提供科学依据。
3. 中药注射剂再评价的方法中药注射剂再评价的方法包括体外实验研究、动物实验研究和临床实验研究。
在体外实验研究方面,可以通过药物化学分析、质量标准研究等方法对中药注射剂进行深入研究。
在动物实验研究方面,可以通过动物药理学实验、动物药代动力学实验等方法对中药注射剂的作用机制和药代动力学特点进行研究。
在临床实验研究方面,可以通过对患者进行临床观察和疗效评价等方法来评估中药注射剂的临床疗效和安全性。
4. 中药注射剂再评价的意义中药注射剂再评价的意义在于提高中药注射剂的临床应用水平,优化中药注射剂的配方和制剂工艺,提高中药注射剂的质量和安全性。
通过中药注射剂再评价的研究,可以对中药注射剂的疗效和安全性进行全面评估,为中医药的发展和临床应用提供科学依据。
个人观点和理解:中药注射剂再评价是推动中药注射剂发展的必要步骤之一。
在现代科技条件下,采用全面系统的方法对中药注射剂进行再评价,可以更好地了解其药理学特点、作用机制、疗效和安全性,进而促进其在临床应用中的规范化和科学化。
中药注射剂再评价也对于中医药的国际化发展具有重要意义,可以提高其在国际上的影响力和竞争力。
四川省中药注射剂安全性再评价工作方案

四川省中药注射剂安全性再评价工作方案一、背景和目的中药注射剂作为一类具有特殊用途的中药制剂,因其疗效显著、使用方便等优点而在临床上得到了广泛应用。
然而,随着中药注射剂的使用量逐年增加,一些严重的不良反应和安全性问题也逐渐凸显出来。
因此,为了保障患者的用药安全,有必要对四川省中药注射剂进行安全性再评价工作。
本次工作旨在通过对四川省中药注射剂的安全性进行再评价,全面了解其不良反应和安全性问题,为临床合理用药提供科学参考依据。
二、工作内容1.数据收集:对四川省各级医疗机构的中药注射剂使用情况进行调查,收集患者用药记录、不良反应报告等相关数据。
2.不良反应分析:对收集到的不良反应报告进行统计和分析,了解不良反应的类型、发生情况、严重程度等。
3.安全性评价:根据不良反应分析的结果和药理学等相关知识,评价中药注射剂的安全性,并给出相应的建议和措施。
4.经验总结和问题分析:对工作中遇到的问题进行总结和分析,提出解决方案,以提高工作效率和准确性。
5.编制报告和推广措施:根据工作内容和评价结果,编制安全性再评价报告,并提出相关的推广措施,以指导临床合理用药。
三、工作步骤1.制定调查方案:明确调查的目的、范围、方法和要求。
2.数据收集:组织专业人员对四川省各级医疗机构的中药注射剂使用情况进行调查,收集患者用药记录、不良反应报告等相关数据。
3.不良反应统计和分析:对收集到的不良反应报告进行统计和分析,了解不良反应的类型、发生情况、严重程度等。
4.安全性评价:根据不良反应分析的结果和药理学等相关知识,评价中药注射剂的安全性。
可以采用临床试验、动物实验、文献分析等方法进行评价。
5.建议和措施提供:根据安全性评价的结果,提出相关的建议和措施,如适应症、禁忌症、用药剂量等,以指导临床合理用药。
6.经验总结和问题分析:对工作中遇到的问题进行总结和分析,提出解决方案,以提高工作效率和准确性。
7.编制报告和推广措施:根据工作内容和评价结果,编制安全性再评价报告,并提出相关的推广措施,以指导临床合理用药。
中药注射剂安全性再评价资料报送要求
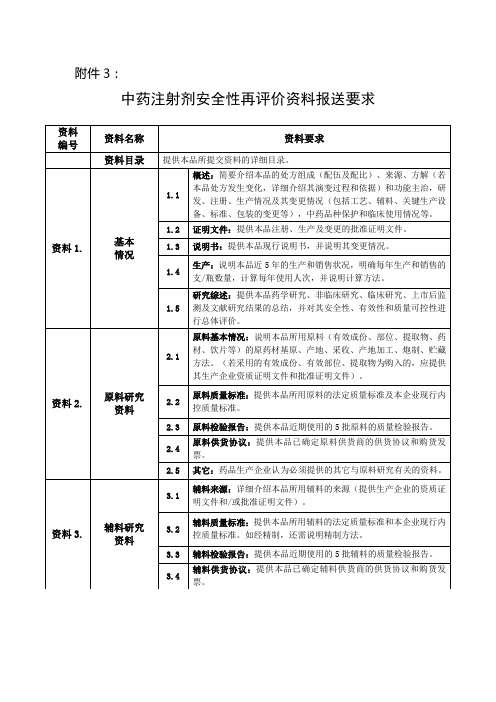
附件3:
中药注射剂安全性再评价资料报送要求
备注:
1. 所有资料用A4纸打印,资料1-12独立装订并报送2份;资料1-8独立装订并报送3份;资料1,4独立装订并报送1份;所有报送资料都应分别准备目录。
2. 在资料首页应注明资料编号名称、单位名称、签章、联系人和联系电话。
3.所有研究资料均需有研究单位公章。
4.已完成的研究资料应保证真实性、完整性。
尚未完成的和未进行的研究应书面予以说明。
5.药品生产企业应一次性提交再评价资料,必要时,可以要求药品生产企业再补充资料。
注射剂一致性评价与无菌药品包装系统密封性创新验证

验证方法,由于检测效率低、精度不高、验证过程可能直接污染药品等诸多原因,在大规模应用时受到了制约。
楚天科技股份有限公司(以下简称楚天科技)研发的无损、高精度的高压放电法、激光顶空分析法,可良好应用于无菌药品包装容器密封性验证,帮助制药企业规避传统方法的弊端和风险,从而顺利通过注射剂一致性评价。
高压放电法
就基本原理而言,高压放电
图1 安瓿高压放电检漏原理示意图
图2 替代法示意图
流程工业
图3 激光在包装上钻孔
完整性方法的检测最小微孔。
此
种方法的缺点在于:成本高,验证
精度受制于激光钻孔与检测手段
的最大能力。
高压放电法验证包装容器密
封完整性
随着密封性检测技术的不断
发展,高压放电法也越来越多地
应用于生产过程中。
相比色水法、
微生物挑战法等传统方法,它具
图4 楚天科技AILM80型灯检检漏一体机
图5 TDLAS激光检测技术原理图
2020-04 制药业47
图6 多种因素致使瓶体内氧气浓度发生变化
图7 1atm氮气下1mL安瓿不同漏孔泄漏速率
图8 激光检测与视觉检测相融合
制药业2020-04。
CDE老师带你梳理注射剂一致性评价中药学要求

CDE老师带你梳理注射剂一致性评价中药学要求仿制药(generic drug)是指具有与原研药品相同的活性成份、剂型、规格、适应证、给药途径和用法用量的原料药及其制剂,仿制药应与原研药品质量和疗效一致,可替代原研药品发挥相同的临床疗效。
仿制药在我国的临床用药中占主导地位,但由于历史的原因,我国早期批准的部分仿制药与原研药品确实存在一定的差距。
随着经济技术的高速发展,国内制药工业研发和制造水平提高,监管意识和能力增强,广大人民群众对优质药品的需求日益迫切,国务院于2012年1月正式印发《国家药品安全“十二五”规划》,提出对已上市的化学仿制药进行质量和疗效一致性评价(以下简称“一致性评价”)。
根据《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》,化学药品新注册分类实施前批准上市的仿制药,包括国产仿制药、进口仿制药和原研药品地产化品种,均须开展一致性评价。
一致性评价将加速我国化药仿制药的技术要求与国际接轨,是提高我国仿制药质量的一项重要工作。
注射剂(Injection)系指药物与适宜辅料制成的供注入机体内的无菌制剂,主要包括注射液、注射用无菌粉末和注射用浓溶液等。
因其直接注射入血管、组织或器官,吸收快,作用迅速,特别是静脉注射的注射剂,药物直接进入血液循环发挥药效,是临床使用中风险较高的剂型,其研发和生产的技术要求也相应更为严格。
本文从参比制剂选择、处方工艺研究、质量研究与控制、包材和稳定性研究等方面,结合国内外指导原则和相关政策文件,分析一致性评价要求下化药注射剂仿制药开发药学要求的变化。
需要指出的是,本文仅就普通注射剂进行讨论,特殊注射剂(如脂质体、胶束、微球、混悬型注射液、静脉乳剂等)还应结合其剂型特点和临床用法用量等进一步评价。
一、参比制剂选择参比制剂是仿制药研发的标杆,选择参比制剂是仿制药研发的开始,参比制剂选择是否正确决定仿制药的成败。
《药品注册管理办法(局令第28号)》定义仿制药为国内批准上市的已有国家标准的药品,在此阶段,原研品的地位没有得到充分的重视,监管机构没有对参比制剂进行统一规定。
2008年第7号文国家食品药品监督管理局关于发布化学药品注射剂和多组分生化药注射剂基本技术要求的通知

国家食品药品监督管理局关于发布化学药品注射剂和多组分生化药注射剂基本技术要求的通知(国食药监注[2008]7号)【收藏】各省、自治区、直辖市食品药品监督管理局(药品监督管理局):为落实国家局制定的《整顿和规范药品研制、生产、流通秩序工作方案》(国食药监办〔2006〕465号),严格审评审批化学药品注射剂、中药注射剂和多组分生化注射剂等安全性风险较大的3类品种。
国家局组织制定了《化学药品注射剂基本技术要求(试行)》和《多组分生化药注射剂基本技术要求(试行)》(以下称《技术要求》),现予发布,请参照执行,并将有关事宜通知如下:一、国家局已受理但尚未批准注册的化学药品注射剂和多组分生化药注射剂应参照《技术要求》进行研究。
二、已经批准注册的化学药品注射剂和多组分生化药注射剂也应参照《技术要求》进行相关研究,并在申报再注册时提供相关研究资料。
三、对已上市化学药品注射剂、多组分生化药注射剂进行仿制、改变剂型或者改变给药途径研究时,研究者应当慎重考虑已上市品种的研究基础。
附件:1.化学药品注射剂基本技术要求(试行)2.多组分生化药注射剂基本技术要求(试行)国家食品药品监督管理局二○○八年一月十日附件1:化学药品注射剂基本技术要求(试行)本技术要求适用于化学药品中各种注册分类的注射剂。
本技术要求主要针对目前化学药品注射剂研发、生产和使用中存在的突出问题,在遵循一般评价原则的基础上,通过分析可能影响注射剂临床使用安全性的主要因素,结合品种的上市基础等,提出化学药品注射剂审评中的重点关注点和相应的技术要求。
一、化学药品注射剂剂型选择的必要性、合理性(一)选择注射途径给药剂型的必要性、合理性对剂型的必要性、合理性进行评价通常应综合考虑如下因素:1.药物的理化性质、稳定性和生物学特性药物的理化性质(溶解度、pKa、分配系数、吸湿性、晶型等)、稳定性(对光、湿、热的稳定性,固、液状态下的稳定性和配伍稳定性)和生物学特性(吸收、分布、代谢、消除等)可以为剂型的选择提供指导,在有些情况下甚至可能限定剂型的选择。
中药注射剂安全性再评价质量控制评价技术原则(试行)
附件2:中药注射剂安全性再评价质量控制评价技术原则(试行)根据《关于开展中药注射剂安全性再评价工作的通知》要求及《中药注射剂安全性再评价质量控制要点》、《中药注射剂安全性再评价基本技术要求》的相关规定,为了更好地开展中药注射剂再评价工作,制定本技术原则。
质量可控是中药注射剂安全有效的基础和保障。
中药注射剂的质量控制工作包括与中药注射剂相关的所有质量控制工作,如原辅料、药材前处理、制备工艺、包装、贮藏、运输、使用等环节的质量控制等,其质量可控性评价应包括与之相关的全过程的质量控制工作。
中药注射剂再评价的质量可控性评价将依据对药学研究资料的评价意见、评价性抽验的报告、生产工艺现场核查结果等作出。
本技术原则明确了对药学研究资料中有关质量研究、质量标准研究及稳定性研究资料的一般要求和基本评价,其所遵循的基本原则和目标是保证质量均一稳定、控制已知的安全风险。
在实际工作中,应注意根据品种特点和具体情况进行有针对性的工作。
一、质量研究评价注射剂的质量研究是指根据工艺、质量标准和稳定性研究的需要而进行的基础研究。
通过质量研究,建立全面系统的质量与风险控制体系和质量保证体系,保证产品质量稳定均一、安全有效。
应根据注射剂质量控制的需要,进行大类成份分析等基础研究,建立合理的检测项目和检测方法,并对产品质量进行检测。
(一)质量研究包含文献研究、化学成份研究、定性定量分析方法研究、生物学质控方法的研究等。
(二)注射剂中所含成份应基本清楚。
应对注射剂总固体中所含成份进行系统的化学研究,明确总固体中所含大类成份的种类及占总固体的量。
有效成份制成的注射剂,其单一成份的含量应不少于90%,多成份制成的注射剂结构明确成份的含量因品种而异。
(三)应结合产品的安全性、有效性及均一性,进行相关质控方法的研究。
二、质量标准研究评价应根据注射剂质量控制的需要,结合质量研究情况,建立合理的检测项目和检测方法,完善和提高质量标准。
(一)质控项目的设置应考虑到注射给药以及中药注射剂自身的特点,并能尽可能全面地、灵敏地反映药品质量的变化情况。
中药注射剂安全性再评价技术要求药学部分--田恒康.
生产工艺评价技术原则
生产工艺 生产工艺不得与法定质量标准的【制法】相违背,否则
应提供相关的批准证明文件。 质量标准【制法】中未明确的工艺参数应在实际生产范
围内细化固定。
生产工艺评价技术原则
生产工艺 提供完整的工艺规程。描述完整的制备工艺,包括工艺路
线、方法及工艺参数等; 提供生产工艺各单元操作(如提取、浓缩、纯化、配液、
中药注射剂存在的问题
关于暂停使用和审批鱼腥草注射液等7个注射剂的通告
国食药监安[2006]218号
国家药品不良反应监测中心病例报告统计表明,使用鱼腥草注射 液等7 个注射剂(见附件)后引起过敏性休克、全身过敏反应、胸 闷、心悸、呼吸困难和重症药疹等严重不良反应,已明确显示该类 药品存在临床用药安全隐患。
检索国家食品药品监督管理局基础数据库,截至2005年11月21日,我 国获准上市的葛根素注射剂包括葛根素氯化钠注射液、葛根素葡萄糖 注射液、葛根素注射液、注射用葛根素四个品种,涉及181个批准文 号。
2004年11月,国家食品药品监督管理局发布了“关于修订葛根素注射 剂说明书的通知”。通报发布后,在国家药品不良反应监测中心病例 报告数据库中,有关葛根素注射剂的新发不良反应病例报告共1006例 (发生时间为2003年1月1日-2005年6月30日);其中,发生时间为 2005年1月1日-6月30日的243例(要求修订说明书通知发出后)。 1006例病例报告中严重不良反应报告30例,其中11例死亡。严重不良 反应报告以急性血管内溶血为主,共18例,其中8例死亡(占死亡病 例的73%),
生产工艺评价技术原则
原料 采取有效措施保证原料质量的稳定 药材的基原、药用部位 产地 采收期 产地加工 贮存条件 包装
关于做好中药注射剂安全性再评价工作的通知
法规准则关于做好中药注射剂安全性再评价工作的通知国食药监办[2009]359号各省、自治区、直辖市食品药品监督管理局(药品监督管理局):为全面提高中药注射剂的安全性、有效性和质量可控性,国家局下发了 关于开展中药注射剂安全性再评价工作的通知 (国食药监办 2009 28号)。
为进一步控制中药注射剂安全风险、做好安全性再评价工作,现就有关事项通知如下:一、全面开展生产及质量控制环节的风险排查,切实控制中药注射剂安全隐患为提高中药注射剂的生产及质量控制水平,国家局组织制定了 中药注射剂安全性再评价质量控制要点 (附件1)(以下简称 质量控制要点 )。
中药注射剂生产企业必须对照 质量控制要点 要求,全面排查本企业在药品生产质量控制方面存在的问题和安全风险,主动采取有效措施,切实控制安全风险,提高产品质量。
中药注射剂生产企业要强化对原辅料供应商的审计,加强对制剂稳定性、产品批间一致性的研究工作,要特别注意对热原、无菌和无效高分子物质控制的自我检查,并开展关键工艺的验证工作,保证产品质量。
企业经自查不能控制产品质量风险的,应立即主动停产,或主动注销药品批准证明文件。
自查结束后,中药注射剂生产企业应将自查整改结果报所在地省级药品监督管理部门。
中药注射剂生产企业应指定专门机构或人员,负责药品不良反应报告和监测工作,对发生的药品不良反应和质量投诉,要及时分析调查,发现存在安全隐患的药品应主动召回,确保临床用药安全。
中药注射剂生产企业应当按照 药品说明书和标签管理规定 (局令第24号)的要求,结合卫生部、国家食品药品监管局、国家中医药局 关于进一步加强中药注射剂生产和临床使用管理的通知 (卫医政发 2008 71号),尽快完善药品说明书的用法、不良反应、注意事项和配伍禁忌等项内容,指导临床合理用药,降低临床使用风险。
各省(区、市)药品监督管理部门要积极组织本辖区中药注射剂生产企业做好生产及质量控制环节的风险排查工作。
中药注射剂再评价指导原则
中药注射剂再评价指导原则
中药注射剂再评价指导原则通常涉及药品质量、安全性、有效性等多个方面,主要目的是确保药品在再评价过程中符合相关的法规和标准。
以下是可能包含在中药注射剂再评价指导原则中的一些关键方面:
1.质量再评价:
•包括对原始生产批次的药品进行质量评估,确保药品的质量符合药典或相关标准。
•对关键质量特性(如成分含量、纯度、微生物限度等)进行再评估。
2.安全性再评价:
•对原始生产批次的药品进行安全性评估,包括药品中可能存在的毒性成分、可能的不良反应等方面的再评估。
•考虑可能存在的新的药物相互作用、禁忌症等信息。
3.有效性再评价:
•对原始生产批次的药品进行有效性评估,确保药品的治疗效果符合预期。
•考虑可能存在的新的疗效信息、患者反应等。
4.生产工艺再评价:
•对原始生产工艺进行再评估,确保其能够稳定生产合格的药品。
•考虑可能存在的新的生产技术、工艺变更等。
5.稳定性再评价:
•对药品的稳定性进行再评估,确保其在规定的保存条件下能够保持质量和有效性。
•考虑可能存在的新的稳定性信息、保存条件变更等。
6.法规遵从再评价:
•确保药品的生产和再评价过程符合相关的法规和标准,包括药品管理法规、GMP(Good Manufacturing Practice)
等。
中药注射剂再评价的具体原则可能根据具体的国家法规、行业标准和具体药品而有所不同。
在进行再评价时,通常需要依赖专业的药品监管机构、药品生产企业以及独立的质量控制和评价机构。
此外,随着时间的推移,再评价的指导原则可能会进行更新和调整,因此建议参考最新的法规和行业标准。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
众
林
机
公司宗旨:帮助客户解决包装泄漏引起的产品 质量问题,帮助客户在‘包装完整性’层面通 过国际认证(FDA、欧盟)
电
设 备
有
包装无损检漏技术的国内开拓者,致力于引进 国外先进的检漏技术
限
公 司
3
包装泄漏引起的产品召回事件
产品 AMO 完全多用途溶 液 咪达唑仑注射液 召回公司 雅培医疗光学有限 公司 Hospira公司 召回日期 2010年7月28日 2010年6月29日 原因
注射器针筒有裂缝
袋子泄漏
2,948,741 注射 器
518,376 袋 5,513 箱 (美国); 353 箱 (国际)
上
2009年11月12日, TPN袋子漏液 2009年11月17日
微孔、封口缺陷、裂缝是导致产品召回的3大原因
上
储存运输环 境
海
众
林
机
封口/轧盖 工艺
包装之间碰 撞
设 备
有
密封组件自 身缺陷
在线测试可以提供更大的保证,可以产生即时的反馈。 缺点是检测精度差,设备停机对生产运行有影响。
上
海
众
林
机
电
设 备
有
限
公 司
上
海
仪器的确认包括评估仪器的功能
众
林
The qualification of instruments or equipment to be used for leak testing includes: 1) evaluation of instrument/equipment functionality, and 2) determination of test system detection capabilities using appropriate calibration tools or reference standards to simulate with-leak test conditions.
限
顶盖缺陷导致泄漏,使得无 菌被破坏
34,629 个
482 个 70,000 个 13,698 瓶
机
电
2011年2月15日
2010年11月11日
海
众
0.9% 氯化钠注射液 TPN全
林
自动注射器
Amgen公司
2009年9月14日, 2010年1月18日
2011年3月4日, 2011年3月23日
首选确定性方法
上
海
众
林
机
电
设 备
有
限
公 司
USP 1207.1 P6
关于方法选择
USP 1207.1 P8
and seal quality test option compatible with the product– package. Off-line testing can accommodate slower test cycle times; for methods in which test time is a performance factor, a slower off-line test is often more sensitive than its on-line counterpart. Off-line tests typically are less costly to perform as they utilize bench-top or smaller scale equipment without the sophisticated product-handling machinery required to support higher speed on-line processes.
林
机
USP 38-1207.2 包装完整性泄漏测试技术
电
USP 38-1207.1 包装完整性和测试方法选择
设 备
USP 38-1207
无菌产品包装完整性评估
有
限
美国药典USP 1207
公 司
10
方法类别及检漏精度级别
高压放电 激光 ● 质量提取 确定性 压力衰减 方法 示 踪 气 法 --● 抽真空模式 真空衰减 水检法 微生物挑战 概率性 示 踪 气 法 --方法 嗅探模式 液体示踪法
有
P9
有效性的项目。若注射剂处方中含有抗氧剂、抑菌剂等
限
公 司
8
中国仿制药一致性评价
上
海
众
林
机
真空衰减方法标准ASTM F2338-09
电
欧盟GMP指南及修订稿
设 备
美国FDA 2008指南
有
美国药典USP 1207
限
公 司
9
国外法规
上
海
众
USP 38-1207.3 包装封口质量测试方法
密封组件的 匹配度
限
电
公 司
包装泄漏原因分析
生产工艺, 如冻干/灭 菌工艺
生产设备故 障
上 海 众 林 机 电 设 备
一致性评价法规
有
限
公 司
P3 • 直接接触药品的内包材的除热原验证或供应商出 • 包装密封性验证,方法需经适当的验证;
上
海
众
• 包材密封性验证,方法需经适当的验证;
林
失败后需要采取的措施;
机
电
设 备
有
限
USP 1207.1 P8
公 司
关于设备确认
USP 1207.1 P9
上
海
精密度是方法能产生可靠、可重复数据的能力
众
林
“Precision” is the ability of the method to yield reliable, repeatable data. Precision includes repeatability (e.g., repeat testing of a homogeneous test sample population), ruggedness (within laboratory tests performed, for example, by multiple operators on multiple days, using multiple instruments; also known as intermediate precision), and reproducibility (among laboratories tests).
机
P4 • 培养基灌装/模拟验证,并明确规定培养基灌装
电
设 备
具的相关证明资料;
有
限
公 司
7
中国仿制药一致性评价
上
海
众
林
(例如压力/真空衰减等)进行检测,并进行方法学验证。
机
容器密封性替代。容器的密封性可采用物理完整测试方法
电
稳定性考察初期和末期进行无菌检查,其他时间点可采用
设 备
辅料,在稳定性研究中还要考察这些辅料含量的变化情况。
上
海
众
离线测试精度更高,设备价格更低
林
机
电
设 备
有
限
公 司
关于方法选择
USP 1207.1 P8 A prerequisite for an on-line leak test method for entire lot testing is that it be nondestructive to the package and its contents. On-line testing can potentially provide greater assurance that all packages have integrity and can yield instant feedback in the event of package misassembly or breakage, enabling real-time line corrections. For some test systems, incorporating large-scale leak detection equipment into a complex high-speed product–package filling and assembly line can be prohibitive. Higher line speeds leading to shorter leak detection test cycle times can limit test method detection capability. In addition, the impact of instrument downtime on the production run as a result of leaking packages or possible equipment malfunction is an important consideration.
上海众林机电设备有限公司
上
海
众
林
机
电
设 备
有
限
公 司
与注射剂再评价相关的 包装完整性评估
上
海
众
林
包装完整性测试方法选择及标准制定
机
电
国外法规
设 备
一致性评价法规
有
限
引言
公 司
提纲
上海众林公司简介
成立于2005年,位于上海市闵行区
上
海
主营3大业务:实验室无损检漏设备、生产线高 速在线检漏机、一致性评价‘包装完整性’测 试服务。