(完整word版)MDD医疗器械指令
(完整word版)Essential Requirements Checklist of the MDD(07 47 EC)基本要求检查表(中英文)
eliminate or reduce risks as far as possible (inherently safe design and construction),
尽可能地消除或降低风险(固有安全设计和结构)
where appropriate take adequate protection measures including alarms if necessary, in relation to risks that cannot be eliminated,
Ok / Fail
符合/不符合
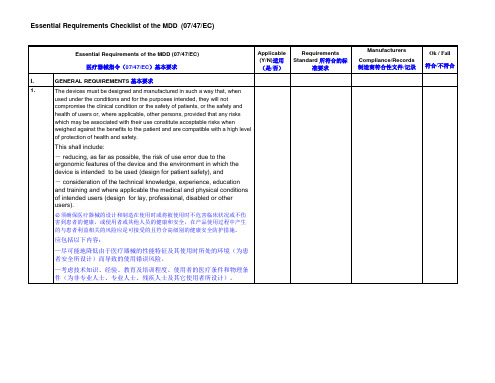
I.
GENERAL REQUIREMENTS基本要求
1.
The devices must be designed and manufactured in such a way that, when used under the conditions and for the purposes intended, they will not compromise the clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, providedthat any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety.
医疗器械CE认证及MDD指令介绍

10
深圳市龙德生物科技有限公司版权所有
医疗器械全球咨询
医疗器械全球贸易
欧盟医疗器械指令
Hlongmed
在欧盟,Council Directive 93/42/EEC of 14 June 1993(简称MDD 指令)中对医疗器械的上市审批与市场监督等做了全面规定。
“Directive(指令)”是欧盟的一种法规类别,欧盟各成员国均必须将指 令的要求转换为各自的国内法律。 MDD指令,是在1993年7月12日公布于Official Journal of the European Communities,并分别在1998年、2000年、2002年和 2003年、2007年进行修订,其中2007年修订案在2010年3月实施。 其中一共有23个条款和12个附录。
6
深圳市龙德生物科技有限公司版权所有
医疗器械全球咨询
医疗器械全球贸易
术语
Hlongmed
“custom-made device” 指依照合格医疗从业人员描述的特色而特别制作的器械,该器械是为特 定病患设计且专供该病患使用。 前述描述可以由专业资格而获授权的其他人提供。 但订制的器械不包括那些为满足医疗人员或其他专业使用人要求而改装 且大量生产的器械。
15
深圳市龙德生物科技有限公司版权所有
医疗器械全球咨询
医疗器械全球贸易
欧盟医疗器械指令
Hlongmed
基本要求的具体包括如下14条: 1、器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安 全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人 们所接受,但应具有高水平的防护办法。 2、生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应 : 首先应尽可能降低甚至避免危险 其次,对无法避免的危险采取适当的防护措施,包括安装报警装置; 最后,告知用户所提供防护措施的弱点及其可能带来的危险。 3、器械必须取得生产者期望获得的功能。器械设计制造和包装应有利于第一条(2)(A )D多规定的各项功能的发挥。 4、在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所 指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康 。 5、器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有 关规定不会发生有不良反应。 6、副作用的大小与器械的使用性能相比较是否可以为人们所接受
MDD-医疗器械指令解读

产品分类
III类 可吸收缝合线, 心脏瓣膜 等
TÜV产品服务
25
TÜV南德意志集团
产品分类
• 分类要点 -- 分类规则的应用由器械的预期使用目的决定
PRODUCT SERVICE
-- 如果器械是和其他器械配合使用,分类规则分别适
用于每种器械附件
--可以和其他一起使用的器械分开独自分类 启动或影响某种器械的软件与器械本身属于同一
条款3.
条款4. 研究
用器械被使用而无需带有CE标志
TÜV产品服务
19
TÜV南德意志集团
医疗器械指令:基本框架
条款5. 条款8. 符合协调标准的医疗器械被认为满足基本要求
PRODUCT SERVICE
如果发现医疗器械不安全的话,保护条款允许某个 成员国采取行动
条款9.
符合性评价程序与产品的分类(附录IX中给出的分 类规则) 有关
9
TÜV产品服务
TÜV南德意志集团
欧洲共同体新方法指令介绍
4. 新方法指令的实施情况
PRODUCT SERVICE
--
已批准24个新方法指令,如
•
• • •
玩具安全指令 (88/378/EEC)
机械设备指令 (98/37/EEC) 人身保护设备 (93/68/EEC) 医疗器械 (93/42/EEC) 等等
TÜV产品服务
6
TÜV南德意志集团
欧洲共同体新方法指令介绍
2. 新方法指令的产生及其作用
PRODUCT SERVICE
b.
80年代之前,欧洲共同体已颁布了近300个协调指令
,缺陷在于:
--
指令的内容过于具体,不仅涉及到具体产品,甚
(完整word版)MDD医疗器械指令

(完整word版)MDD医疗器械指令医疗器械指令(MDD)93/42/EEC 简介什么是医疗器械?“医疗器械”是指制造商预定用于人体以下目的的任何仪器、装置、器具、材料或其他物无论它们是单独使用还是组合使用,包括为其正常使用所需的软件:疾病的诊断、预防、监视、治疗或减轻;损伤或残障的诊断、监视、治疗、减轻或修补;解剖学或生理过程的探查,替换或变更;妊娠的控制医疗器械的评估等级:所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE 标志,以说明产品符合欧盟制定的相关指令。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。
它根据不同的要求共分为6个等级,供认证机构评估。
设计阶段生产阶段I级自我符合声明自我符合声明I级(测量功能)自我符合声明申报机构I级(灭菌)自我符合声明申报机构IIa级自我符合声明申报机构IIb级申报机构申报机构III级申报机构申报机构认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系。
由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。
医疗器械的风险分析:EN1441失效模式及后果分析(FMEA);失效树分析(FTA);上市后的监控(客户投诉情况等);临床经验根据EN1441风险分析的一些例子;器械的预期用途;预期与病人和第三者的接触;有关在器械中所使用的材料/元件的风险;供给患者或来自患者的能量;在无菌条件下生产的器械;用于改变病人环境的器械;说明用器械;用于控制其它器械或药品或与其配合使用的器械;不需要的能量或物质的输出;易受环境影响的器械;带有重要消耗品或附件的器械;必要的日常维护和校正;含有软件的器械;货架寿命有限制的器械;延迟或长期使用可能造成的影响;普通风险;所有的适用项目必须论述包括可能的危险和降低风险的方法。
医疗器材产品安全指令MDD

医疗器材产品安全指令MDD医疗器材产品安全指令(Medical Device Directive,简称MDD)于1993年颁布,是欧洲联盟针对医疗器材和设备所制定的指令,旨在保证医疗器材产品的安全性和性能稳定性,以保障公众的健康和安全。
MDD的实施是为了将欧盟成员国之间的医疗器材市场整合,建立起一个相互认可的法规体系,以便医疗器材能够在整个欧洲联盟范围内自由流通。
MDD赋予了制造商有关其产品负责的义务,包括确保产品符合相关的安全和有效性要求,并进行整个生命周期的管理。
这些要求包括产品的设计、制造、包装、技术文件和申报等方面。
同时,MDD为监管机构提供了相应的权力和职责,用以监督和评估医疗器材的合规性。
此外,MDD还规定了符合条件的非欧盟制造商可以通过授权代表的形式来进入欧洲市场。
MDD要求医疗器材制造商必须进行严格的质量管理体系,并确保其产品符合相关标准和指南。
制造商需要对其产品进行全面的技术评价,并申报其产品的合规性。
这些评估必须基于标准化的测试和验证方法,以确保产品在各种使用环境和条件下的安全性和性能。
此外,制造商还必须建立和保持技术文件,这些文件包括产品的技术规格、设计文件、制造过程控制和质量管理等。
MDD还规定了欧洲联盟各成员国的监管机构的角色和职责。
这些机构负责审核技术文件、进行市场监督和进行必要的抽样和检测。
此外,监管机构还负责对产品进行风险评估和分类,并发布有关产品的安全警告和召回信息。
总体而言,医疗器材产品安全指令MDD在欧洲联盟范围内确保了医疗器材产品的安全性和性能可靠性,促使制造商建立和维护质量管理体系,并提供相关的技术文件和申报。
监管机构监督和评估医疗器材的合规性,确保产品在市场上的安全和有效性。
通过这一指令,欧洲联盟建立起了一个统一的医疗器材市场,有助于促进医疗器材的自由流通,提高医疗质量和安全水平。
MDD指令

MDD 93/42/EEC号指令1993年6月14日理事会第93/42/EEC号指令关于医疗器材欧洲共同体理事会,依据欧洲经济体所制订的罗马条约,特别是第100a条规定,依据执委会的建议案,配合欧洲议会,依据经济暨社会委员会的意见。
1.鉴于内部市场之完成应采取一些措施,鉴于内部市场是一无内部疆界之区域,区域内之货品、人员、服务及资金应可自由流通。
2.鉴于各会员国间现存有关医疗器材之安全,对健康之保护及使用特性方面之法律、法规及行政命令之内容与范围不尽相同,鉴于各会员国对此器材之验证及检验程序也不相同,鉴于前述之分歧将阻碍共同体内的贸易活动。
3.鉴于医疗器械之使用对病患、使用者、甚至其他人有关安全及健康保护的相关国家规定应加以调和,以保证此类器材在内部市场能自由流通。
4.鉴于调和之规定必然与各会员国采取之部分措施有所不同,些措施是为筹措公共健康与疾病保险计划之基金,且直接或间接与医疗器材有关,于共同体若与上述措施相符,则这些规定并不影响会员国落实上述措施的能力.5.鉴于医疗器材应提供病患、及第三者高度之保护,应该达到厂商所要求之性能水准,于维持或改进各会员国对病患等保护的程度乃本指令目的之一。
6.鉴于部分医疗器材是符合1965年1月26日理事会第65/65/EEC号指令,与专卖医药产品有关之法律、法规或管理行为所订之实施规定,鉴于医疗器材之上市基本上由本指令规范,但医疗产品之上市则受65/65/EEC号指令规范,鉴于若有某种器材须与其他医疗产品组成一完整的产品而上市销售,使用,且无法二次使用时,则该组合产品应受65/65/EEC号指令规范。
鉴于前述之医疗器材和包含医疗物质且该物质单独使用时符合65/65/EEC号指令规定之医疗器材应加以区别。
鉴于前述包含于医疗器材之医疗物质若对人体产生作用以辅助医疗器材之作用时,则该医疗器材之上市应由本指令规范,鉴于1975年5月20日75/318/EEC号理事会指令[制定各会员国在测试专利医疗产品方面有关分析药物毒性和临床之标准及调查书的法律调和],医疗物质之安全、品质及效用在前述情况下则须依该指令明定之适当方法加以证实。
MDD指令规则

MDD指令范围93/42/EEC规则 1非插入式器械属于I类器械,但适用以下其他规则的出外。
规则 2用于输送和储存血液、体液或人体组织和其他液体和气体为人体吸收、服用或注入的非插入式样器械,属于IIa类:·如果他们可以同IIa或更高类别的有源器械连接使用;或·如果他们的预定功能属于储存和输送血液和其他体液、储存人体器官或人体组织。
其他的都属于I类。
规则 3插入式器械用于改变血液、体液和其他注入人体的液体的生物和化学成分属于III类器械。
但处理方法属于过滤、气体和热能的分离或交换的该类非插入式器械属于IIa。
除此以外的其他情况属于I类。
规则 4同受伤皮肤接触的非插入式器械:·如果用于形成机械屏障,阻止或吸收渗出液体,属于I类;·如果主要用于辅助治疗已经伤及真皮的创伤,属于Iib类;·其他情况属于IIa,包括主要用于处理创伤周围环境的器械。
规则 5插入式器械,除非属于外科手术插入式器械或同有源器械连接使用,·如果属于暂时性的使用方式,属于I类;·如果属于短期性的使用方式,属于IIa类;但不包括在口腔中仅至咽部、在耳道中仅至耳鼓、在鼻腔中使用但不被粘膜吸收的器械;·如果属于长期性的使用方式,属于IIb类;但在口腔中仅至咽部、在耳道中仅至耳鼓、在鼻腔中使用但不被粘膜吸收的器械属于IIa类;其他需要与IIa或更高类别的器械连接使用的插入式器械,属于IIa类;但不包括外科手术插入式器械。
规则 6暂时性使用方式的外科手术插入式器械属于IIa类,但以下情况除外:·如果为了诊断、监测或矫正心脏或主血管系统疾病,器械直接触及这些器官,在属于III类;·可重复使用的外科器械属于I类;·以电离辐射的方式提供活力(energy)的器械属于IIb类;·对人体生理发生作用或为人体全部或大部分所吸收的器械属于IIb类;·通过发送装置给人体施用药物,如果对人体具有某种危险,则属于IIb类。
医疗器械MDD介绍

医疗器械MDD介绍医疗器械MDD(Medical Device Directive)是欧洲联盟对医疗器械监管的指导性法规。
MDD于1993年开始实施,并在2024年被新的医疗器械法规(MDR)取代。
尽管如此,MDD的理念和原则仍然对医疗器械的管理和监管具有重要影响,并为理解和应用MDR提供了基础。
MDD的主要目的是确保医疗器械在欧洲市场上的安全性和有效性,同时促进欧洲国家的技术创新和市场竞争。
MDD适用于用于医疗目的的任何设备、仪器、工具、设施和系统(无论是否为人体使用),以及有关诊断、预防、监测、治疗或缓解疾病的装置、程序、材料或其他物品。
MDD的适用范围包括各类医疗器械,例如体温计、手术器械、人工关节等。
根据MDD的分类规定,医疗器械被分为四个类别,从高风险到低风险依次是:Ⅰ类、Ⅱa类、Ⅱb类和Ⅲ类。
每个类别的医疗器械需要遵守一定的法规要求,并进行相应的评估程序,如CE认证。
此外,MDD还对医疗器械的设计和生产、技术文件和质量控制、市场准入申请等方面提出了详细的规定。
根据MDD的要求,医疗器械制造商需要满足一系列的法规要求,例如确保其产品质量符合要求、设立质量管理体系、进行技术文件的编制和评估、获得合格的认证机构的审查等。
此外,制造商还需要负责跟踪和报告产品在市场使用过程中出现的任何事故和负面事件。
MDD还详细规定了与医疗器械相关的其他利益相关方的责任和义务。
例如,MDD强调医疗器械的使用者和操作人员需要对其使用医疗器械的正确方法进行培训,并确保医疗器械的正确使用和维护。
此外,MDD还规定了医疗机构需要建立医疗器械的追溯能力和不良事件报告系统,以及卫生监管机构和认证机构的监管职责和职能。
总的来说,医疗器械MDD是欧洲对医疗器械的管理和监管指导性法规,旨在保证医疗器械的安全性和有效性,并促进欧洲市场的创新和竞争。
MDD对医疗器械的分类、设计和生产、市场准入申请等方面提出了具体要求,要求制造商遵守相应的法规,并进行相应的评估程序。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
医疗器械指令(MDD)93/42/EEC 简介
什么是医疗器械?
“医疗器械”是指制造商预定用于人体以下目的的任何仪器、装置、器具、材料或其他物
无论它们是单独使用还是组合使用,包括为其正常使用所需的软件:
疾病的诊断、预防、监视、治疗或减轻;
损伤或残障的诊断、监视、治疗、减轻或修补;
解剖学或生理过程的探查,替换或变更;
妊娠的控制
医疗器械的评估等级:
所有进入欧盟市场的产品,企业必须具有表示自我符合声明的CE标志,以说明产品符合欧盟制定的相关指令。
医疗器械指令(MDD),MDD指令适用于大多数进入欧盟销售的医疗设备。
它根据不同的要求共分为6个等级,供认证机构评估。
设计阶段生产阶段
I级自我符合声明自我符合声明
I级(测量功能)自我符合声明申报机构
I级(灭菌)自我符合声明申报机构
IIa级自我符合声明申报机构
IIb级申报机构申报机构
III级申报机构申报机构
认证机构的统一评估包括根据指令规定的基本要求评审技术文件、根据标准EN 46001 或EN/ISO 13485评审质量体系。
由于美国、加拿大和欧洲普遍以ISO 9001, EN 46001或ISO 13485作为质量保证体系的要求,故建议质量保证体系的建立均以这些标准为基础。
医疗器械的风险分析:
EN1441
失效模式及后果分析(FMEA);
失效树分析(FTA);
上市后的监控(客户投诉情况等);
临床经验
根据EN1441风险分析的一些例子;
器械的预期用途;
预期与病人和第三者的接触;
有关在器械中所使用的材料/元件的风险;
供给患者或来自患者的能量;
在无菌条件下生产的器械;
用于改变病人环境的器械;
说明用器械;
用于控制其它器械或药品或与其配合使用的器械;
不需要的能量或物质的输出;
易受环境影响的器械;
带有重要消耗品或附件的器械;
必要的日常维护和校正;
含有软件的器械;
货架寿命有限制的器械;
延迟或长期使用可能造成的影响;
普通风险;
所有的适用项目必须论述包括可能的危险和降低风险的方法。
医疗器械CE认证基本要求:
基本要求是MDD的最重要部分,它包括所有的医疗器械通用要求:
一、基本要求(总要求)
安全性(任何风险与器械提供的益处相比较,必须在可以接受的范围内,故亦称风险分析);
风险的可预防性或被消除性,至少应给予警告(报警系统或警戒报警系统);
性能符合性(产品的基本要求);
器械性能和安全的效期(器械的安全和性能必须在器械的使用寿命内得到保证。
);
器械的储存和运输(应保证器械在合理的运输、储存条件下不受影响)。
二、基本要求的具体包括如下14条
器械设计和生产必须保证:按照其预定和条件使用,器械不会损害医疗环境、患者安全、操作者或其他人员的安全和健康;使用时的潜在危险与患者受益相比较可以为人们所接受,但应具有高水平的防护办法。
生产者的设计和制造方案,必须考虑在现有工艺技术条件下遵守安全准则、生产者应:首先应尽可能降低甚至避免危险;其次,对无法避免的危险采取适当的防护措施,包
括安装报警装置;最后,告知用户所提供防护措施的弱点及其可能带来的危险。
器械必须取得生产者期望获得的功能。
器械设计制造和包装应有利于第一条(2)(A)D多规定的各项功能的发挥。
在生产线者确定的器械使用寿命期内,在正常使用可能出现的压力,第1、2、3款所指的各项性能应保持稳定,不能危害医疗环境、危害患者、使用者或其他人员的健康。
器械的设计、生产和包装应当保证,器械的性能在运输和储存过程中只要遵守有关规定不会发生根本逆变。
副作用的大小同器械的使用性能相比较可以为人们所接受。
化学、物理和生物性能。
感染和微生物污染。
组装和环境因素。
检测器械。
辐射防护。
带有能源或与其他能源相连接的器械。
生产者提供的操作信息。
如果需要根据医疗数据确定器械是否满足基本要求,如第六款的情形,有关数据必须按照MDD附录Ⅹ的规定取得。
三、CE标志-医疗器械指令(MDD)TCF文件内容
医疗设备CE认证技术档案所需内容:
生产商/或欧洲代表名址;
产品及型号描述;
EC符合声明书;
风险评估;
基本安全点检表;
适用之调合标准/或其他标准;
市场反馈及抱怨分析;
使用说明及标签;
授权代表;
线路、图表(适用的话);
计算书、测试报告或其它证明材料;
检验过程及过程描述;
灭菌或其它特殊过程(适用的话);
灭菌类产品的包装材料及方法;
质量体系、质量手册;。