diamond_3_教程系列1.01
最新Diamond基础操作指南(大全)
Diamond基础操作手册Crystal Impact Diamond是一款分子和晶体结构可视化软件。
Diamond整合了丰富的功能,可以简化处理晶体结构数据冗长的工作,不仅适用于研究和教学,同时也可以应用与出版和作报告。
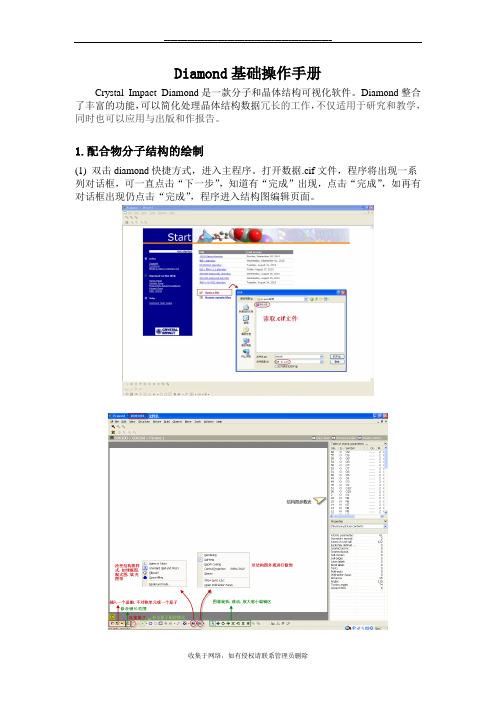
1.配合物分子结构的绘制(1) 双击diamond快捷方式,进入主程序。
打开数据.cif文件,程序将出现一系列对话框,可一直点击“下一步”,知道有“完成”出现,点击“完成”,如再有对话框出现仍点击“完成”,程序进入结构图编辑页面。
(2) 在做图之前,首先点击修改键长按钮,出现键长对话框,将不可能成键的原子组去掉(如Cd-C Cd-H),并根据已知键长范围设定原子组,如下图Cd-O键最大值约在2.7,所以将计算机默认值由2.880改为2.700。
(3) 选择添加不对称单元原子按钮,将在屏幕上出现该配合物一个不对称单元中的原子(未键连)。
(4) 在Build菜单中找到Filter,单击出现对话框,在该对话框中,原子前若挑钩则表示其在屏幕中会正常显示出来。
如果不挑钩则表示该原子被过滤掉,将不显示出来。
在配合物的结构图中一般H原子不需要显示出来,所以我们将H原子的钩去掉。
(5)配位环境图的绘制。
选择屏幕左下方的连接原子按钮,将(3)中孤立的原子连接起来。
由于配位环境图的绘制需要补全与金属离子键合的所有分子,因此使用补全配位模式按钮,将另外两个三唑长出来。
对于配位环境图中的非碳原子需标出原子序号,方法为鼠标左键单击要编号原子,然后选择屏幕下方的原子标号按钮。
如果对于程序直接给出的原子标号字体大小不满意,可以鼠标左键双击原子标号进入原子标号设定对话框进行设定。
注意对后长出来的与金属离子直接相连的原子需要根据该原子的对称操作符号(与.txt文件中的对称操作对应)给出相应的对称操作记号。
如N2的对称操作为x,-y+1/2,z+1/2 在.txt文件中对应的操作码符号为#1,则该原子在标注时为N2A。
Diamond 3 界面选项的功能介绍

在本章中我们首先介绍一下一个典型的 diamond 3 界面上所有选项的基本功能。
一个典型的 diamond 3 的界面 打开 C:\Program Files\Diamond 3\Tutorial\文件夹中的 pyrene.cif 文件(上图所示)。
第一节 File 菜单系列 1.1 File 菜单简介
图 3.2 Navigation 效果图
图 3.3 Navigation 选项 Navigation 后在左侧显示出了各个菜单的名称主要包括五项。 ☻点击 Structure x 可以显示当前操作的结构图:
图 3.4 点击 Structure x 效果 ☻点击 Data sheet 可以显示该结构的晶体学信息:
表 3-2 Distance/angles 操作界面说明
可以选择参与列表的原子种类 Unit
表示选择范围的单位,?表示埃,下面依次是皮米和纳
米
Dmin Dmax
表示选定的原子该范围球壳范围内的所有原子,默认值
angle
d 1,2 and 1,3
Count
Symmetry op. Atom code Coordinate Standard Uncertainties (s.u.值)
File 菜单系列,包括 Windows 系统常规的几种选项(如图 2 所 示 。)
图 1.2 File 菜单系列 1.2 常用选项 1.2.1Open 选项
点击该选项,可以看到 diamond 3 可以打开的所有文件类型(如图 3 所 示 )。
图 1.3 Diamond 3 支持的阅读格式
前三项是该公司开发的 Diamond 及 Endeavour 软件的默认格式。其中 Cif 文件格式最 为通用。ICSD/Crystin 及 CSD-FDat 是两个晶体学数据库输出的文件格式。Protein Data Bank 格式表示支持蛋白质晶体数据库文件。常用的格式还包括笛卡儿 xyz 座标格式,这在构建特 殊结构模型时极为便利,比如我们会在后面章节中提到的螺旋体的构建。 1.2.2 Save 选项
POV-Ray与Diamond3联用作图简介

POV-Ray与Diamond 3 联用作图简介第一节POV-Ray基本知识POV-Ray,全名是Persistence of Vision Raytracer,是一个使用光线跟踪绘制三维图像的开放源代码免费软件。
很多漂亮的图片就是由POV-ray来制作的。
本文的主要目的在于介绍如何利用Diamond软件联合POV-Ray 画图,我们不可能将所有的功能加以介绍。
本节简要介绍一下。
1.坐标系统POV-Ray是基于Ray-tracing的渲染软件,学习它的第一个事情便是认识其坐标系统。
2. POV-Ray文件的基本构成和很多计算机语言一样,文件格式中有很多属于专用语言,这些文字在程序中往往表现为彩色。
一个基本的POV-Ray文件有如下几个部分:一些头文件、摄像机定义、场景物体定义、灯光定义。
a. 头文件,常用的包括:#include "colors.inc" // 定义基本颜色#include "shapes.inc" // 定义基本形状物体#include "textures.inc" // 定义一些物体属性注://后面的文字是注释,不参与程序运行。
b. 摄像机位置和观察方向当用POV-Ray的标记语言来表示一个摄像机的时候,我们可以这样来写:camera {location <0, 2, -3> //(大家可以想象摄像机在坐标中的位置)look_at <0, -1, 2> //(摄像机的位置确定后,还要让镜头对着某个方向)up x*1 //(up和right定义了纵横视口比例)right y*1}正规写法如下:camera {location <0.0, 2.0, -3.0>look_at <0.0, -1.0, 2.0>right x*image_width/image_height}c. 主要物体比如我们要在<0, 0, 0>位置(原点)放置一个半径为2黄色的球体可以表示为(注意{}总是成对出现,color是系统默认关键字,黄色若想成为关键字,被默认为黄色,应首字母大写):sphere {<0, 0, 0>, 2texture {pigment {color Yellow} }}到此,我们完成了所有工作,就可以渲染(run)了,但您看到的将是一个黑色背景上的一个很淡的黄色的实心圆。
Diamond 软件的使用

这就是DIAMOND,一款在原子水平实现晶体结构可视化 的软件能为您做到的。
LOGO
Diamond
介绍
1.用户界面介绍 2.看图工具介绍
LOGO
软件界面
LOGO
用户界面
导航栏 表 格 面 板
LOGO
测量工具栏
LOGO
转换工具栏
LOGO
视频工具栏
LOGO
起步阶段
如何导入 晶体结构 数据文件
如何通DIAMOND自动 绘制晶体结构图
Diamond 软件的使用
LOGO
Edit your company slogan
内容
1、软件的作用
2、diamond介绍
3、起步阶段
4、绘制图像
5、实 例
LOGO
学习 Diamond 软件的作用?
如果你有晶体结构数据(空间群参数,单胞参数,原子参 数或者晶体数据文件,如cif文件),而且你希望, 1.绘制用于演示文稿或者出版物的高质量图片? 2.了解晶体结构的构造原理? 3.通过各种方式向客户展现晶体构造原理?
图像视角
LOGO
实 例
LOGO
LOGO
起步阶段
如何显示晶体结构数据、 距离和角度表格 以及粉末衍射图样
LOGO
绘制图像
如何手动输 入晶体结构 数据
绘制图像
如何通过结 构图绘制助 手)绘制图 像
LOGO
如何手动输入晶体结构数据
新建空 白文档
打开新 建结构 助手
输入晶胞参 数和空间群
输入原子参数 (元素、氧化 态以及配位数)
完成数据输入 ,进入结构图 像绘制助手
Diamond基础操作指南(大全)
Diamond根底操作手册Crystal Impact Diamond是一款分子和晶体结构可视化软件。
Diamond 整合了丰富的功能,可以简化处理晶体结构数据冗长的工作,不仅适用于研究和教学,同时也可以应用与出版和作报告。
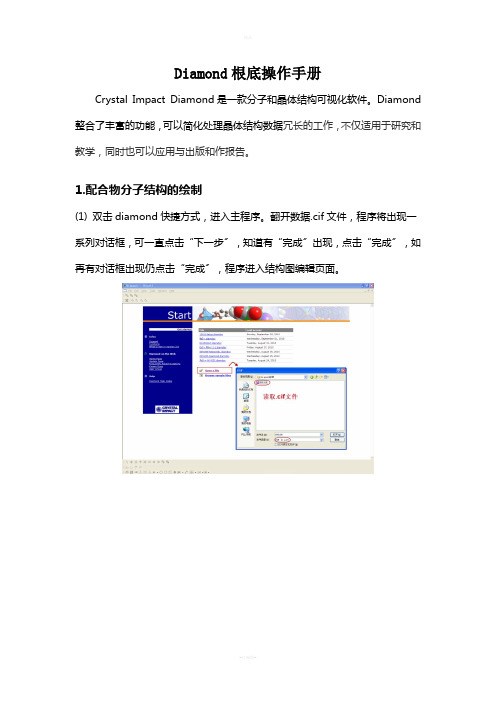
1.配合物分子结构的绘制(1) 双击diamond快捷方式,进入主程序。
翻开数据.cif文件,程序将出现一系列对话框,可一直点击“下一步〞,知道有“完成〞出现,点击“完成〞,如再有对话框出现仍点击“完成〞,程序进入结构图编辑页面。
(2) 在做图之前,首先点击修改键长按钮,出现键长对话框,将不可能成键的原子组去掉〔如Cd-C Cd-H〕,并根据键长范围设定原子组,如下列图Cd-O键最大值约在2.7,所以将计算机默认值由2.880改为2.700。
(3) 选择添加不对称单元原子按钮,将在屏幕上出现该配合物一个不对称单元中的原子〔未键连〕。
(4) 在Build菜单中找到Filter,单击出现对话框,在该对话框中,原子前假设挑钩那么表示其在屏幕中会正常显示出来。
如果不挑钩那么表示该原子被过滤掉,将不显示出来。
在配合物的结构图中一般H原子不需要显示出来,所以我们将H原子的钩去掉。
(5)配位环境图的绘制。
选择屏幕左下方的连接原子按钮,将(3)中孤立的原子连接起来。
由于配位环境图的绘制需要补全与金属离子键合的所有分子,因此使用补全配位模式按钮,将另外两个三唑长出来。
对于配位环境图中的非碳原子需标出原子序号,方法为鼠标左键单击要编号原子,然后选择屏幕下方的原子标号按钮。
如果对于程序直接给出的原子标号字体大小不满意,可以鼠标左键双击原子标号进入原子标号设定对话框进行设定。
注意对后长出来的与金属离子直接相连的原子需要根据该原子的对称操作符号〔与.txt文件中的对称操作对应〕给出相应的对称操作记号。
如N2的对称操作为x,-y+1/2,z+1/2 在.txt文件中对应的操作码符号为#1,那么该原子在标注时为N2A。
晶体结构立体模型建构软件-Diamond的使用指南

晶体结构立体模型建构软件-Diamond的使用吴平伟中国海洋大学材料科学与工程研究院E-mail: wupingwei@晶体结构立体模型建构软件-Diamond的使用在使用Diamond软件构造晶体模型时,需要知道晶体的结构数据,即晶体的空间群、晶胞参数和原子坐标。
晶体结构数据可以手动输入,也可以直接从晶体信息文件中获得。
我们将通过几个例子来说明软件的使用方法。
一、NaCl晶体结构模型的构造下面我们以NaCl为例手动输入晶体结构数据。
NaCl晶体的结构数据为:空间群Fm-3m(225);晶胞参数a=5.64Å;原子坐标Na:4a, Cl:4b。
我们将通过这个例子学会如下操作:1、学会手动输入晶体结构数据;2、学会晶体模型的构造;3、学会旋转晶体模型,从不同的角度观察;4、学会改变背景和原子及晶胞的颜色等参数;5、学会以一种原子为中心,另一种原子为配位原子构造配位多面体;6、学会多面体外观的设计。
打开软件,界面如下图所示:点击“File| New”,出现一对话窗口,如下图,选择第二个选项,按“OK”。
结果生成一个名字为Diamond1的空白的页面,同时弹出一个名字为New Structure的对话窗口,点“下一步”,在新弹出的窗口中确认Crystal Structure with cell and Spacegroup被选中,在Cell length中输入5.64,如下图:注意Space group(空间群)后是否我们需要的NaCl晶体的空间群Fm-3m(225),如果不是,点击Browse钮,在弹出的对话窗口中选中Fm-3m(225),即在Fm-3m(225)上点击使其变蓝色,如下图。
点“OK”回到前面的对话窗口。
点“下一步”(在出现的如下图的对话框中可以输入原子坐标,即在“Atomic parameters“中输入相应的元素符号和原子坐标值,但我们将在其他的地方做这个工作)点“下一步”,在出现的Completing the new structure Assistant窗口中有三个选项:Start structure picture; Launch the structure picture creation assistant;Create structure picture automaticly。
diamond-3-教程系列1.01

GB50057—1994 建筑物防雷设计规范(2000年版)
GB50343—2004 建筑物电子信息系统防雷设计规范
GB 50303—2002 建筑电气工程施工质量验收规范
GB 50074—2002 石油库设计规范
GB 50156—2002(2006 年版)汽车加油加气站设计与施工规范
GB50169-2006电气装置安装工程接地装置安装及验收规范
13.8
将不同的电气装置、导电物体等,用接地导体或浪涌保护器以某种方式连接起来,以减小雷电流在它们之间产生的电位差。
13.9
建筑物需要规定和控制雷击电磁脉冲环境的区域,可划分为LPZ0A、LPZ0B、LPZ1……LPZn+1区。
13.10
也称电涌保护器,用于限制暂态过电压和分流浪涌电流的装置,它至少应包含一个非线性电压限制元件。
一、检查并记录电涌保护器数量、型号、安装位置及工艺,各级电涌保护器的主要技术参数应符合设计规范要求,电涌保护器装置应有具备检测资格的部门出具的检测报告或主管机构颁发的《防雷产品使用许可证》。
二、电涌保护器的标志应完整和清晰,表面应平整、光洁、无划伤、无裂痕和烧灼痕或变形。有劣化显示及工作状态指示的电涌保护器其状态指示应与生产厂家产品说明相一致。没有劣化显示及工作状态指示的电涌保护器,应检查电涌保护器表面手感温度,当接近或大于人体温度时,应更换电涌保护器。
Diamond基础操作指南(大全)
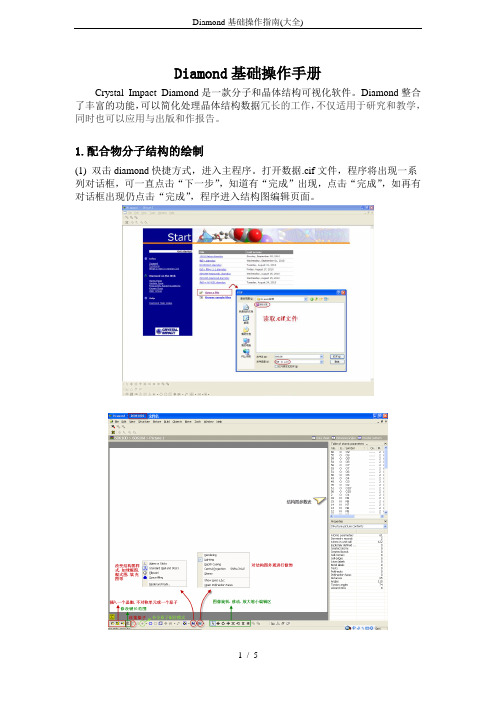
(4)在Build菜单中找到Filter,单击出现对话框,在该对话框中,原子前若挑钩则表示其在屏幕中会正常显示出来。如果不挑钩则表示该原子被过滤掉,将不显示出来。在配合物的结构图中一般H原子不需要显示出来,所以我们将H原子的钩去掉。
(5)配位环境图的绘制。选择屏幕左下方的连接原子按钮,将(3)中孤立的原子连接起来。由于配位环境图的绘制需要补全与金属离子键合的所有分子,因此使用补全配位模式按钮,将另外两个三唑长出来。对于配位环境图中的非碳原子需标出原子序号,方法为鼠标左键单击要编号原子,然后选择屏幕下方的原子标号按钮。如果对于程序直接给出的原子标号字体大小不满意,可以鼠标左键双击原子标号进入原子标号设定对话框进行设定。注意对后长出来的与金属离子直接相连的原子需要根据该原子的对称操作符号(与.txt文件中的对称操作对应)给出相应的对称操作记号。如N2的对称操作为x,-y+1/2,z+1/2在.txt文件中对应的操作码符号为#1,则该原子在标注时为N2A。
1.配合物分子结构的绘制
(1)双击diamond快捷方式,进入主程序。打开数据.cif文件,程序将出现一系列对话框,可一直点击“下一步”,知道有“完成”出现,点击“完”,如再有对话框出现仍点击“完成”,程序进入结构图编辑页面。
(2)在做图之前,首先点击修改键长按钮,出现键长对话框,将不可能成键的原子组去掉(如Cd-C Cd-H),并根据已知键长范围设定原子组,如下图Cd-O键最大值约在2.7,所以将计算机默认值由2.880改为2.700。
(6)原子颜色与键长的修改,在picture菜单下的Atom Design和Bond Design选项。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
第一章Diamond 3 界面选项的功能介绍在本章中我们首先介绍一下一个典型的diamond 3界面上所有选项的基本功能。
一个典型的diamond 3 的界面打开C:\Program Files\Diamond 3\Tutorial\文件夹中的pyrene.cif文件(上图所示)。
第一节File菜单系列1.1 File菜单简介File菜单系列,包括Windows系统常规的几种选项(如图2 所示。
)图1.2 File菜单系列1.2 常用选项1.2.1Open 选项点击该选项,可以看到diamond 3 可以打开的所有文件类型(如图3所示)。
图1.3 Diamond 3 支持的阅读格式前三项是该公司开发的Diamond 及Endeavour软件的默认格式。
其中Cif文件格式最为通用。
ICSD/Crystin及CSD-FDat是两个晶体学数据库输出的文件格式。
Protein Data Bank 格式表示支持蛋白质晶体数据库文件。
常用的格式还包括笛卡儿xyz座标格式,这在构建特殊结构模型时极为便利,比如我们会在后面章节中提到的螺旋体的构建。
1.2.2 Save 选项这里默认的保存格式是Diamond 3 Document (*.diamdoc)格式。
1.2.3 Save as选项共包括三个次级选项:图1.4 Diamond 3 Save as的三个次级选项在实际应用中,前两项功能相似,我们以Save Document As为例进行介绍:Save Document As提供14种文件格式:图1.5 Diamond 3 支持的14结构储存格式前三项为该公司开发的结构文件格式,常用的为第一项*.diamdoc。
通常在我们处理一个较为复杂的结构时,一次无法完成或者以后仍需要修改时,必须保存成该格式。
该格式详细保留了您的一切设置(分子模型的模式、原子的半径颜色、键长等),demo版不提供该格式的保存。
其它格式则通常并不实用。
需要指出的是,有时为了统计自建模型(比如在抽象拓扑结构时,统计两种或多种拓扑类型的比例)中原子的个数及比例,可以保存为cif格式。
图1.6 Diamond 3 支持的6种图片储存格式Save Graphic As:本功能提供了图片保存功能,共包含了6种图片格式。
我们通常需要使用的是bmp格式(位图格式,文件通常较大,在word中缩放会导致分辨率降低)、jpg格式(文件较小,在win office软件中可以任意缩放而不改变分辨率)、tif格式(是很多杂志要求的图片格式,易于编辑和处理)。
为实现diamond与3D Max软件的交叉使用,还需要掌握另外一种文件保存格式,即wrl格式,该格式也保留了创作者构建的所有信息,而不只是结构单元。
第二节Edit菜单系列2.1 Edit菜单简介Edit菜单主要包括结构及其模式类型的选择、复制和粘贴图2.1 Edit菜单主要选项2.2 常用功能2.2.1 Undo与Redo典型的windows界面选项,不需要过多说明,只是要注意,在计算机内存较小而图片由异常复杂时,可能造成死机。
2.2.2 Copy及Paste 选项本功能提供了两种功能(1)复制屏幕所显示的图片,可以粘贴在其它文档中,比如常用的office文档中。
图2.2 Copy 选项的结构复制功能(2)复制屏幕显示的结构包括各种详细信息,粘贴到另一个打开的diamond 3 文档中,该功能可以实现结构的对接,但是必须要注意的是不同结构可能由于空间群的不同无法实现对接,这是可以采用如下的方法:新建一个空文档>>打开C60结构文件:调解两个窗口的大小以适合显示:直接复制C60结构,粘贴到pyrene文档中,无法实现,相反的复制-粘贴过程也被警告无法进行。
提示如下(图2.3):图2.3 结构复制时对称操作信息必须考虑图2.4结构复制时消除对称操作通过对pyrene文档Structure>>Remove Translational Symmetry(图2.4)消除了对称操作后,选择,复制并粘贴C60结构,得到如下结果:图2.5 结构的对接很明显,这时新的结构并没有任何对称操作,需要增加对称操作。
那么,反过来,将消除了对称操作的pyrene粘贴到C60文档中,仍然不被允许。
对接的技巧,读者可以自己试验,这样不必在设计结构时一个一个原子的输入,大大节省了我们的时间。
Paste 选项仅仅能实现结构的粘贴,并不能实现外部数据(如图片或结构)的导入。
2.2.3 Copy Style及Paste Style选项:该选项可以实现将某些设定好的类型选项直接通过类似Word 中格式刷的功能进行类型更换。
操作起来可以首先选择某个原子或键,然后点击Copy Style,然后再选择一些原子或键,点击Paste Style:图2.6 右键点选下的Copy Style操作图2.7 Paste Style操作图2.8 Copy>>Paste Style操作后的效果由于我们在Paste Style操作中选择了部分H原子,使得这部分的原子的颜色和半径都和碳原子一样,但仍需注意,Style的复制粘贴只限于原子或键的格式,并不能复制粘贴原子的种类,换句话说,那些氢原子格式发生了改变,但仍是氢原子。
这两种功能在细化处理时很有效,不必一个一个原子的去设置,同时又避免了大面积选择时的失误操作。
2.2.4 Select系列操作:Select all 就不必介绍了;Invert Selection 是常用的功能,比如我们在处理一个很复杂的结构时,需要改变其中一个较大部分的类型,很明显,Select all无法实现,而一一选择费时且在diamond中目前仍必须按住Ctrl,才可以复选或多选原子,数目太多,容易前功尽弃,这时,我们可以通过先选择较少部分,然后Invert Selection来实现。
Select Molecule(s) 该功能中所说的分子并不等同于在其它软件中的分子。
本软件中,只要成键,就被认为是分子中的一部分,同时,一个分子中的部分如果键被打断则被认为是不同分子。
这种功能非常实用,尤其是在配位聚合物的结构分析中。
图2.8例图如图2.8中所示的红色键,是人为加入的一条键,当左键点击选择相互连接的两个分子时,程序认为这其实是一个分子。
图2.9 右键点选下的Select Molecule(s)操作图2.10 Select Molecule(s)操作效果图该功能的快捷操作还可以用鼠标右键中的Select Molecule(s)选项:图2.11 左键点选下的Select Molecule(s)快捷操作Lasso Selection 该功能与Photoshop、Chemdraw的功能非常相似,可以实现精挑细选,而不像按住左键进行方框选择功能那样笨拙,操作时,和其它软件一样,选择完毕后必须回到出发点才结束一次选择。
图2.12 右键点选下的Lasso Selection操作图2.13 Lasso Selection操作轨迹图第三节View 系列菜单3.1 View 系列菜单简介该系列菜单包含了所有“查看”信息,包括操作文档的大纲及缩略图、晶体学参数及列表、原子及键的信息列表(其中还可以进行选择)、粉末图显示、数据/图片切换等功能。
3.1 典型的View 操作界面3.2 常用选项介绍3.2.1 Navigation 和Thumbnails这两项功能分别提供了目录(大纲)和缩略图功能。
图3.2 Navigation效果图图3.3 Navigation选项Navigation后在左侧显示出了各个菜单的名称主要包括五项。
☻点击Structure x 可以显示当前操作的结构图:图3.4点击Structure x效果☻点击Data sheet可以显示该结构的晶体学信息:图3.5 Data sheet 列表信息这些信息可以打印或直接拷贝到其它文档处理软件,并直接列表,这些信息可以直接用于文章的发表。
比如,选择-复制-拷贝到本教程后的信息如下:表3-1 Data sheet 列表信息GeneralOriginCode Structure 1Database datesCommon nameSystematic nameStructural formulaAnalytical formulaBibliographic dataAuthor(s)Publication titleCitationMineral nameCompound sourceStructure typeCreation method Created with Diamond v2.0CommentsPhase dataFormula sum H40 C64Formula weight 809.02 g/molCrystal system monoclinicSpace-group P 1 21/a 1 (14)Cell parameters a=12.3027 Å b=9.9879 Å c=8.2206 Å β=96.40°Cell ratio a/b=1.2318 b/c=1.2150 c/a=0.6682Cell volume 1003.83 Å3ZCalc. density 1.33821 g/cm3Meas. densityMelting pointRAllRObsPearson code mP104Formula type N5O8Wyckoff sequence e26Atomic parameters ,Anisotropic displacement parameters, in Å2列表限于篇幅,不再列出。
☻点击Distance/angles,则给出一格相当详细的信息表。
表3-2 Distance/angles操作界面说明Select atom (type)s可以选择参与列表的原子种类Unit表示选择范围的单位,?表示埃,下面依次是皮米和纳米DminDmax表示选定的原子该范围球壳范围内的所有原子,默认值是0-2.5Å。
angle 该选项提供了在设定球壳内与之相关的原子所组成的夹角,具体操作见后面说明。
d 1,2 and 1,3 该选项只能与angle选项共同使用,不能单独选择,列出了在angle选项中构成角度的三个原子间的距离Count 对选型范围内重复出现的原子累计计数,在选择的半径较小时,一般为1×,如果调大Dmax,有的原子可能在该范围内出现多次,表示为2×,3×等。
Symmetry op. 列出对称操作Atom code H10 C14 135550111.0664 红字部分就是原子代码Coordinate 原子坐标Standard Uncertainties 选择该项会使得2.0295数值显示成2.0295(25) ,25就是s.u.值,Angle 使用说明:在前面我们看到(以H10 为例)在H10为原点的0-2.5Å范围内共有六个原子,使用angle功能后,明显六个原子的所有组合被列出,共C 26种情况。