精细有机合成技术第六章
精细有机合成单元反应

精细有机合成单元反应第二章磺化、硫酸化反应1. 什么叫磺化反应?什么叫硫酸化反应?哪些物质可以作磺化剂?三氧化硫硫酸发烟硫酸氯磺酸硫酰氯磺化反应为亲电取代,硫酸磺化时,亲电质点是什么?2. 脂肪饱和烃磺氧化,磺氧化的历程。
3. 硫酸的浓度对磺化反应的影响4. 简述“共沸去水磺化法”5. 写CH 3+SO3生成三种异构体的机理6. 反应式R CH CH 2+NaHSO 3R CHCH 2+H 2SO 4ROH +ClSO 3HClNO 2NO 2+NaHSO 3+MgO60-65℃nahso37. 磺化活性排序浓硫酸、发烟硫酸,SO 3(CH 3)3N ·SO 38. 用硫酸磺化法合成OH加100%硫酸磺化,加NaOH 成盐,加NaOH 成酚钠盐,加硫酸成酚 9. 为什么芳香烃磺化时浓硫酸的浓度越高越容易生成砜?用什么方法可以阻止砜的生成?第三章硝化反应1. 混酸硝化反应机理2. 与硝酸比,混酸硝化法的优点3. 如何除掉硝基化合物中的酚杂质4. 如何去掉NO2NO2由少量的NO2NO2NO2NO25. (烟)H SO 36. 合成OHCH 3OCHO OHCH 3OCHONO 27.CH3亚硝化反应历程第四章 卤化反应1.写出FeCl 3催化氯化芳烃的Mechanism 。
为什么单质碘对芳环氢化有催化作用2.写出次氯酸进行酸催化氢化的历程及特点具有较小的空间阻碍3.FeCl3催化氯化芳烃时,为什么原料的纯度有很大影响4.简述沸腾氯化法流态化氯化又称沸腾氯化。
流态化 简称流化 利用流动流体的作用 将固体颗粒群悬浮起来 而使固体颗粒具有某些流体 表现特征 因此强化了气一固间或液一固间的接触过程。
这种使固体颗粒具有某些流体特征的技术被称为流态化技术。
5.为什么OH在无催化剂存在的条件下,能以很高的速度进行氯化6.Ar-H+Cl2CuClAr-Cl+HCl7.写出芳烃侧链氯化光引发历程?链引发链增长链终止8.+CH2O+HCl2氯甲苯+H2O9.CH3O NH2NaNO2/HBF410℃10.OCH3ICl碘化对甲氧基苯11.OH+PCl5第五章还原反应1. 举例说明芳硝基化合物Fe还原成芳胺后的分离方法。
第六章相转移催化有机合成

第6章相转移催化有机合成§6.1 概论相转移催化是20世纪60年代开始形成的一门新技术,多应用于非均相反应体系,可以在温和的反应条件下加快反应速率,简化操作过程,提高产品收率,受到了人们极大的关注。
相转移催化最初只是应用于烷基化等几类典型的反应中,现已迅速发展到许多化学化工领域。
除了应用于有机合成、高分子聚合反应外,还进入了分析、造纸、制革等领域。
这一新技术的推广,对许多化工新产品的开发、老产品生产技术的改造、质量的提高和成本下降产生了较大的影响,特别是在精细化工和制药方面,将带来令人瞩目的经济效益和社会效益。
6.1.1 相转移催化反应的定义什么是相转移催化有机合成呢?它是指在相转移催化剂作用下,有机相中的反应物与另一相(水相或固体相)中的反应物发生的化学反应,称为相转移催化(Phase Transfer Catalysis,PTC)反应。
例如:PhOH + C4H9Br—→ PhOC4H9+ HBr (6-1)其中苯酚PhOH是固态的,溶于水中。
而溴丁烷是液体,溶于有机溶剂中。
两种反应物不能同时存在于相同的相中进行接触,所以该反应是属于两相间的亲核取代反应。
要完成该反应,可以有以下4种方法:(1)将两种反应物分别溶于水和有机相中,进行强烈搅拌,但所用温度和压力较高,产率低。
(2)将溴丁烷改成丁醇,用浓硫酸进行催化。
但是由于浓硫酸的腐蚀性强,且温度较高(>140℃),所以该方法受到了限制。
(3)用William法合成,即在无水乙醚中,用苯酚钠和C4H9Br直接反应。
但是无水操作较麻烦。
(4)用四丁基溴化铵为相转移催化剂,可在50℃下进行反应,产率>90%。
该方法使用了相转移催化剂,可使两相中的反应物充分接触,且在较低的温度下进行反应,产率较高。
是一种先进的合成方法。
1056.1.2 相转移催化反应的发展过程相转移催化有机合成的历史大约有60年。
从反应类型、反应机理到反应的应用发展很快,已经成为重要的有机合成方法。
有机合成课件6章(缩合反应)(最新版)
CH CH Ph
H3C O2N P2N O NO2
4. Knoevenagel反应: 这类反应的特点是一个亚甲基上连接两个吸电子基团, 使得其氢活性明显提高,反应较易进行。一般使用弱碱(有 机胺)作催化剂即可,甚至不使用催化剂。
第六章 缩合反应
如:
CH2(CN)2
O CH2-C-H
CH3CHO
OH CH3-CH-CH2-CH=O 不稳定,易脱水
CH3CH=CH-CHO
从反应过程来看,应该有两种可能缩合产物。表示如下:
第六章 缩合反应
O CH2-C-H (1) CH3-CH=O CH2=CH-O O H C-CH3 (2) CH3-CH-CH2-CH=O O CH3-CH-CH2-CHO (1) OH H =-16.62KJ/mol
和卤代酸酯的反应(Darzenes) 该反应也要在强作用下完成。如:
O + ClCH2CO2C2H5 t BuOK 83~95% O CO2C2H5
其反应过程为:
ClCH2CO2C2H5 Cl
t BuOK
ClCHCO2C2H5
O
CHCO2C2H5 O O
CO2C2H5
第六章 缩合反应
3,醛(酮)与其它化合物的缩合: 1)硝基化合物:
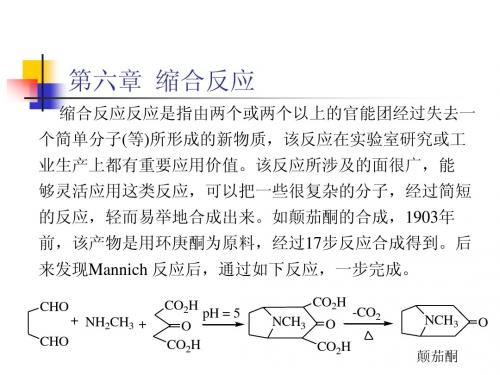
CHO CHO + NH2CH3 + CO2H O CO2H pH = 5 CO2H NCH3 O CO2H -CO2 NCH3 O
颠茄酮
第六章 缩合反应
一,碳负离子历程的缩合反应
这类反应很多,如羟醛(酮) 缩合,酯缩合等。其反应历程为:
C + C= O C O C 产物
1,醇醛缩合:
CH3CHO NaOH -H2O
《精细有机合成及工艺》思考题
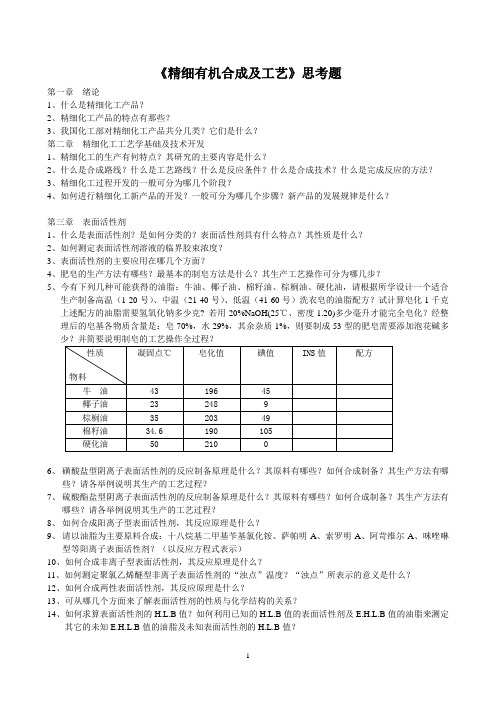
《精细有机合成及工艺》思考题第一章绪论1、什么是精细化工产品?2、精细化工产品的特点有那些?3、我国化工部对精细化工产品共分几类?它们是什么?第二章精细化工工艺学基础及技术开发1、精细化工的生产有何特点?其研究的主要内容是什么?2、什么是合成路线?什么是工艺路线?什么是反应条件?什么是合成技术?什么是完成反应的方法?3、精细化工过程开发的一般可分为哪几个阶段?4、如何进行精细化工新产品的开发?一般可分为哪几个步骤?新产品的发展规律是什么?第三章表面活性剂1、什么是表面活性剂?是如何分类的?表面活性剂具有什么特点?其性质是什么?2、如何测定表面活性剂溶液的临界胶束浓度?3、表面活性剂的主要应用在哪几个方面?4、肥皂的生产方法有哪些?最基本的制皂方法是什么?其生产工艺操作可分为哪几步?5、今有下列几种可能获得的油脂:牛油、椰子油、棉籽油、棕榈油、硬化油,请根据所学设计一个适合生产制备高温(1-20号)、中温(21-40号)、低温(41-60号)洗衣皂的油脂配方?试计算皂化1千克上述配方的油脂需要氢氧化钠多少克? 若用20%NaOH(25℃、密度1.20)多少毫升才能完全皂化?经整理后的皂基各物质含量是:皂70%,水29%,其余杂质1%,则要制成53型的肥皂需要添加泡花碱多6、磺酸盐型阴离子表面活性剂的反应制备原理是什么?其原料有哪些?如何合成制备?其生产方法有哪些?请各举例说明其生产的工艺过程?7、硫酸酯盐型阴离子表面活性剂的反应制备原理是什么?其原料有哪些?如何合成制备?其生产方法有哪些?请各举例说明其生产的工艺过程?8、如何合成阳离子型表面活性剂,其反应原理是什么?9、请以油脂为主要原料合成:十八烷基二甲基苄基氯化铵、萨帕明A、索罗明A、阿苛维尔A、咪唑啉型等阳离子表面活性剂?(以反应方程式表示)10、如何合成非离子型表面活性剂,其反应原理是什么?11、如何测定聚氯乙烯醚型非离子表面活性剂的“浊点”温度?“浊点”所表示的意义是什么?12、如何合成两性表面活性剂,其反应原理是什么?13、可从哪几个方面来了解表面活性剂的性质与化学结构的关系?14、如何求算表面活性剂的H.L.B值?如何利用已知的H.L.B值的表面活性剂及E.H.L.B值的油脂来测定其它的未知E.H.L.B值的油脂及未知表面活性剂的H.L.B值?15、7%的Tween—60(14.9)和Arace—60(2.1)来乳化31.1%的矿物油(E.H.L.B值10.5)及17.5%的硬化油(10)试求出乳化剂的使用比例。
精细有机合成单元反应复习.docx

精细有机合成单元反应复习第一章精细有机合成基础知识要点一、取代基效应1、诱导效应1)因为成键原子的电负性差异而引起的电荷传递作用2)所有取代基都存在诱导效应,即诱导效应无处不在3)了解诱导效应强弱、方向是认识该效应的核心2、共辄效应1)了解共觇效应的类型、能够判断分子或屮间体是否存在共觇效应2)共辘效应影响的结果是因为电子离域或共享而使分子、活性中间体稳定。
如正离子、负离子或自由基都能够被共辘所稳定。
3)共轨效应方向受诱导影响如分析下面分子或活性中间体的共轨方向H2C^=C—C^=CH2H2C^=C—H3C—C CH—c—OC2H5H H H3、空间效应1)空间效应产生原因一般有两种:A因为取代基体积而产生的空间位阻;B因为分子的刚性结构或键角张力2)与诱导效应一样,所有分子都可能存在空间效应3)空间效应对分子或活性中间体的影响可能是起促进或阻碍作用二、有机反应历程1、脂肪族取代反应重点了解亲核取代反应的影响因素2、芳香族亲电取代了解一般反应历程、取代反应的定位是重点(包括多取代基存在下的取代反应、稠环的单取多取代)3、芳香族亲核取代了解影响亲核収代的影响因素特别是环上已有収代基存在下对亲核取代的活性影响三、溶剂1、了解溶剂的分类2、溶剂对反应活性的影响Huges-Ingold规则3、了解非质子传递性溶剂对亲核反应的影响例题:作为亲核试剂,卤素阴离子在一般极性溶剂屮亲核活性顺序如何?在非质子传递性溶剂中亲核性顺序又如何?试对此作出合理解释。
考占.P 八、、•1、収代基效应的判断:下面反应物或活性中间体存在共轨效应的是()2、极性溶剂对下面反应有活化作用的是()3、下面芳香族亲核取代反应活性最高的是()4、下面亲核试剂亲核性最强的是()5、下面自由基稳定性最好的是()6、在水中卤素阴离子亲核性顺序是:在DMF溶剂中亲核性顺序是:解释其原因?7本章习题28、芳香族亲电取代定位效应第二章卤代反应知识要点一、芳香族环上卤代反应1、亲电反应历程、酸催化反应或无需催化2、连串反应(即有多卤代反应),控制反应产物的关键是卤化反应深度3、反应有较弱的可逆性4、反应的一般条件:反应溶剂:反应物本体、有机溶剂、酸、水反应试剂:单质、次卤酸、NCS、NBS、HX+氧化剂反应活性:与反应物有关、与试剂有关、与溶剂有关主要反应:苯卤代、甲苯卤代、苯酚卤代、苯胺卤代5、碘代反应的特殊性二、不饱和桂的加成卤代1、加成反应活性都很高,在温和的条件下都能够实现加成2、加成方向马氏规则(本质上是取代基效应)3、加成试剂HX、X2、HOX4、共觇烯坯的加成1, 2■加成比1, 4■加成活性更高,所以反应可能首先生成1, 2■加成产物,但1, 4■加成产物稳定性更好,较高的反应温度或较反的反应时间,1, 4■加成产物可能会成为主要产物三、烯丙位或侧链的自由基卤代1、反应是自由基历程,反应条件光照或加热,引发剂能够促进反应2、当有多个位置可能被取代时,节位或烯丙位优先3、反应有多取代异构体,由卤化深度的控制4、侧链卤代吋,不能有过渡金属离子存在(即Lewis酸),否则可能有环上卤代发生5、烯丙位卤代反应的竞争是双键的加成,高温不会发生加成,但低温下反应,主要产物是加成产物四、疑基置换反应1、醇的置换卤代反应活性:短链〉长链、叔醇〉仲醇〉伯醇醇〉酚卤代试剂:浓HX酸-浓盐酸、浓蛍澳酸(HI不宜)含磷卤代试剂・PX3、PX5含硫卤代试剂-SOCb2、酚的置换卤代只能用强卤代试剂如PX3、PX.53、酸的卤代制备酰卤卤代试剂一般用SOCB、PX3五、拨基a—位卤代1、反应是指醛、酮、酸衍生物的。
精细有机合成经典题目

精细有机合成经典题目(总9页)-CAL-FENGHAI.-(YICAI)-Company One1-CAL-本页仅作为文档封面,使用请直接删除第二章精细有机合成基础2.1肪族亲核取代反应有哪几种类型?2.2S N1和S N2反应速度的因素有哪些各是怎样影响的2.32.4卤代烷分别与醇钠和酚钠作用生成混醚的反应,为什么通常各在无水和水溶液中进行,且伯卤代物的产率最好,而叔卤代物的产率最差?2.5什么叫试剂的亲核性和碱性二者有何联系2.62.7为什么说碘基是一个好的离去基团,而其负离子又是一个强亲核试剂这种性质在合成上有何应用2.82.9卤代烷和胺在水和乙醇的混合液中进行反应,试根据下列结果,判断哪些属于S N2历程?哪些属于S N1历程?并简述其根据。
2.10 <1> 产物的构型发生翻转; <2> 重排产物不可避免; <3> 增加胺的浓度,反应速度加快; <4> 叔卤代烷的速度大于仲卤代烷; <5> 增加溶剂中水的含量,反应速度明显加快; <6> 反应历程只有一步; <7> 试剂的亲核性越强,反应速度越快。
2.11比较下列化合物进行S N1反应时的速率,并简要说明理由:C2H5CH2BrCH BrC2H53C BrCH33C2H5(2) C6H5CH2Cl (C6H5)2CHCl C6H5CH2CH2Cl(1)2.12比较下列化合物进行S N2反应时的速率,并简要说明理由:<1>C2H5CH2BrCH BrC2H5CH3C BrCH3CH3C2H5BrICl<2>2.13下列每对亲核取代反应中,哪一个反应的速度较快为什么2.142.152H2OCH3CH2CH3CH3CH2CH3CH3CH2CH2OH + NaBrCH3CH2CH2Br + NaOH<1>CH3CH2CH2Br + NaOH CH3CH2CH2OH + NaBrH2OH2O CH3CH2CH2SH + NaBrCH3CH2CH2Br + NaSH<2>O2N CH2Cl + H2O O2N CH2OH + HCl△△CH3CH2OH + HCl CH3CH2Cl + H2O<3>2.16烷和硫脲进行亲核取代后再用碱分解,可以得到硫醇:2.17RX + H22SR S CNH2NH2..++R S CNH2NH2X-RSH + NH2CN2-2.18请解释在硫脲中,为何是硫原子作为亲核质点,而不是氮原子作为亲核质点?2.192.20为什么说在亲核取代反应中消除反应总是不可避免的?2.21什么叫诱导效应其传递有哪些特点2.222.23最常见的共轭体系有哪些?2.24什么叫共轭效应其强度和哪些因素有关传递方式和诱导效应有何不同2.25在芳环的亲电取代反应中,何谓活化基何谓钝化基何谓邻对位定位基何谓间位定位基2.26影响芳环上亲电取代反应邻对位产物比例的因素有哪些如何影响2.272.28写出下列化合物环上氯化时可能生成的主要产物,并指出每一情况中氯化作用比苯本身的氯化,是快还是慢:NHCOCH3OCH3Br COOC2H5CH2CNCCl3C(CH3)3COCH32.29用箭头示出下列化合物在一硝化时主产物中硝基的位置:F CH3CH3NO2CH3CH3CF3C(CH3)3COCH3CNCH3OCH3NO2CH3 NC NHCOCH3SO3HCH3SO2NH22.19有沿键传递,且位置越远影响越弱这一特性的电子效应是_______。
精细有机合成化学以及工艺学 第六章 硝化以及亚硝化

15
6.3.6硝化副反应 (1)主要的硝化副反应:氧化、去烃基、置换、 脱羧、开环、聚合等。其中氧化影响最大,生成 硝基酚。 (2)烷基苯硝化时,硝化液颜色发黑变暗,说明 硝酸用量不足或硝化温度过高。 因 为 形 成 了 络 合 物 (C6H5CH3· 2ONOSO3H· 2SO4)。 3H 破坏方法:45~55℃时补加HNO3或混酸 (3)大多数副反应,与体系中存在的氮的氧化物 有关。
-8
1.210
-8
10
结论:苯环上有给电子基,硝化反应速度快,产物 主要是邻、对位 苯环上有吸电子基,硝化速度减慢,产物主要 是间位。 2.芳烃硝化异构产物 带有吸电子的取代芳烃硝化邻位异构体 的生成量比对位异构体多。原因是硝基易同 邻位取代基中带负电荷的原子形成络合物。
CHO 19 72 9 1.3 COOH 18.5 80.5 2.2 CN 17.1 80.7 1 NO 2 9 90 8.2 40.8 NO 2 51 OCH 3
F . N . A. D.V .S . 1 F . N . A. D.V .S . F . N . A. 100 % 1 D.V .S .
22
6.4.2 混算配制 配酸工艺:水、硫酸、硝酸如何混合? 先将浓硫酸先缓慢、后渐快加入到水中, 在40度以下先慢后快加入硝酸。
23
6.4.3硝化操作
(4)非均相混酸硝化
被硝化物和硝化产物在反应温度下都呈液态,且 难溶于混酸时,常采用非均相的混酸硝化。这时 需剧烈搅拌,使有机相充分地分散到酸相中以完 成硝化反应。
(5)有机溶剂中硝化
可避免大量使用硫酸作溶剂。 (6)气相硝化 NO2与苯于80~190℃通过分子筛处理便转化为 硝基苯。
5
6.2硝化理论解释
《精细有机合成基础》PPT课件(2024版)

I: -CH3是斥电子基 使苯环上电子云密度
+C、+I同向,都使苯环上电子云 密度
15
有-I,无T: 如-N+(CH3)3、-CF3、-CCl3等 (1)使σ-配合物均不稳定,使苯环钝化; (2)使邻、对位取代产物更不稳定; (3)为间位定位基。
16
有-I,-T: 如-NO2、-CN、-COOH、-CHO等 (1)诱导效应与共轭效应作用一致; (2)则使苯环钝化; (3)间位定位基。
℃) 2.47
26
2.1 芳香族亲电取代的定位规律
4、亲电试剂的空间效应(P10 表2-3) 5、新取代基的空间效应 6、反应的可逆性
27
7、反应条件的影响
温度
(1)通常情况下,温度升高,亲电取代反应活性增 高,选择性下降。
混NO酸2 硝化
NO 2
NO 2
NO 2
+
+
NO 2
0℃ 5%
94%
NO 2 NO 2
-I:吸电子 +I:供电子 共轭效应(T):包括π-π共轭和p-π共轭。
-T:吸电子 +T:供电子
14
有+I,无T: 如-C2H5,-CH3 (1)使σ-配合物稳定,活化苯环;
(2)使H 邻、对位取代产物更稳定H ; (3)H 为C 邻H、对位定位基。 H C H
C: s-p超共轭 使苯环上电子云密度
四、苯环上已有两个取代基的定位规律
已有两个取代基为同一 类型定位基,且处于间 位,则定位作用一致。
已有两个取代基为同一 类型定位基,且处于邻、 对位,则定位作用不一 致——取决于定位能力 的强弱。
CH3
少量
COOH
CH3
主产物
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
无水三氯化铝具有很强的吸水性,遇水会立即分解 放出氯化氢和大量热,严重时甚至会引起爆炸;与空气 接触也会吸收其水分水解,并放出氯化氢,同时结块并 失去催化活性。
第二节 烷基化反应的基本原理
因此,无水三氯化铝应装在隔绝空气和耐腐蚀的密 闭容器中,使用时也要注意保持干燥,并要求其他原料 和溶剂以及反应容器都是干燥无水的。
第二节 烷基化反应的基本原理
(2)质子酸 其中主要是氢氟酸、硫酸和磷酸,催 化活性次序如下:
HF> H2SO4> P2O5> H3PO4 、阳离子交换树脂
第二节 烷基化反应的基本原理
工业上生产烷基苯时,通常采用的是AlCl3-盐酸络 合物催化溶液,它由无水三氯化铝、多烷基苯和微量水 配制而成,其色较深,俗称红油。它不溶于烷化产物, 反应后经分离,能循环使用。烷基化时使用这种络合物 催化剂比直接使用三氯化铝要好,副反应少,非常适合 大规模的连续化工业烷基化过程,只要不断补充少量三 氯化铝就能保持稳定的催化活性。
离子对的形式参与反应。仲卤代烷则常以离子络合物的 形式参与反应。
第二节 烷基化反应的基本原理
(3)用醇烷基化的反应历程 当以质子酸作催化剂 时,醇先被质子化,然后解离为烷基正离子和水:
如用无水AlCl3为催化剂,则因醇烷基化生成的水会 分解三氯化铝,所以需用与醇等物质量之比的三氯化铝:
烷基化反应的活泼质点是按下面途径生成的:
磷酸是较缓和的催化剂,无水磷酸(H3PO4)在高 温时能脱水变在焦磷酸。
第二节 烷基化反应的基本原理
工业上使用的磷酸催化剂多是将磷酸沉积在硅藻土、 硅胶或沸石载体上的固体磷酸催化剂,常用于烯烃的气 相催化烷基化。由于磷酸的价格比三氯化铝、硫酸贵得 多,因此限制了它的广泛应用。
阳离子交换树脂也可作为烷基化反应催化剂,其中 最重要的是苯乙烯-二烯乙苯共聚物的磺化物。它是烯烃、 卤烷或醇进行苯酚烷基化反应的有效催化剂。优点是副 反应少,通常不与任何反应物或产物形成络合物,所以 反应后可用简单的过滤即可回收阳离子交换树脂,循环 使用。缺点是使用温度不高,芳烃类有机物能使阳离子 交换树脂发生溶胀,且树脂催化活性失效后不易再生。
RI>RBr>RCl 当卤烷中卤素原子相同,而烷基不同时,反应活性次序 为:
此外,应明确指出,不能用卤代芳烃,如氯苯或溴苯来代替 卤烷,因为连在芳环上的卤素受到共轭效应的稳定作用,其 反应活性较低,不能进行烷基化反应。
第二节 烷基化反应的基本原理
(2)烯烃 烯烃是另一类常用的烷基化剂,由于烯 烃是各类烷化剂中生产成本最低、来源最广的原料,故 广泛用于芳烃、芳胺和酚类的C-烷基化。常用的烯烃有: 乙烯、丙烯、异丁烯以及一些长链α-烯烃,它们是生产 长碳链烷基苯、异丙苯、乙苯等最合理的烷化剂。
第一节 概述
烷基化反应在精细有机合成中是一类极为重要的反应, 其应用广泛,经其合成的产品涉及诸多领域。最早的烷基化 是有机芳烃化合物在催化剂作用下,用卤烷、烯烃等烷化剂 直接将烷基引入到芳环的碳原子上,即所谓C-烷基化 (Friedel-Crafts反应,简称F-C反应)。利用该反应所合成的 苯乙烯、乙苯、异丙苯、十二烷基苯等烃基苯,是塑料、医 药、溶剂、合成洗涤剂的重要原料。通过烷基化反应合成的 醚类、烷基胺是极为重要的有机合成中间体,有些烷基化产 物本身就是药物、染料、香料、催化剂、表面活性剂等功能 产品。如环氧化物烷基化(O-烷基化)可制得重要的聚乙二 醇型非离子表面活性剂。采用卤烷烷化剂进行氨或胺的烷基 化(N-烷基化)合成的季铵盐是重要的阳离子表面活性剂、 相转移催化剂、杀菌剂等。例如,烷基酚聚氧乙烯醚是用途 极为广泛的非离子型表面活性剂(TX-10、OP-10),其可由 C-烷化及O-烷化反应生产。
第二节 烷基化反应的基本原理
(2)C-烷基化是可逆反应 烷基苯在强酸催化剂存 在下能发生烷基的歧化和转移,即苯环上的烷基可以从 一个苯环上转移到另一个苯环上,或从一个位置转移到 另一个位置上,如:
第二节 烷基化反应的基本原理
当苯量不足时,有利于二烷基或多烷基苯的生成; 苯过量时,则有利于发生烷基转移,使多烷基苯向单烷 基苯转化。因此在制备单烷基苯时,可利用这一特性使 副产物多烷基减少,并增加单烷苯总收率。
第二节 烷基化反应的基本原理
3.C-烷基化反应历程 (1)用烯烃烷基化的反应历程 烯烃常用质子酸进 行催化,质子先加成到烯烃分子上形成活泼亲电质点碳 正离子;
用三氯化铝作催化剂时,还必须有少量助催化剂氯 化氢存在。AlCl3先与HCl作用生成络合物,该络合物与 烯烃反应而形成活泼的碳正离子;
第二节 烷基化反应的基本原理
用卤烷作烷化剂时,也可以直接用金属铝作催化剂, 因烷基化反应中生成的氯化氢能与金属铝作用生成三氯 化铝络合物。在分批操作时常用铝丝,连续操作时可用 铝锭或铝球。
第二节 烷基化反应的基本原理
无水三氯化铝能与氯化钠等盐形成复盐,如 AlCl3·NaCl, 其熔点185℃,在140℃开始流体化。若需要 较高的烷化温度(140~250℃)而又无合适溶剂时,可 使用此种复盐,它既是催化剂又是反应介质。
无水氟化氢的活性很高,常温就可使烯烃与苯反应。 氟化氢沸点19.5℃,与有机物的相溶性较差,所以烷基 化时需要注意扩大相接触面积;反应后氟化氢可与有机 物分层而回收,残留在有机物中的少量氟化氢可以加热 蒸出,这样便可使氟化氢循环利用,消耗损失较少。采 用氟化氢作催化剂,不易引起副反应。当使用其他催化 剂而有副反应时,通常改用氟化氢会取得较好效果。但 氟化氢遇水后具有强腐蚀性,其价格较贵,因而限制了 它的应用。目前在工业上主要用于十二烷基苯的合成。
无水三氯化铝是各种F-C反应中使用最广泛的催化剂。它 由金属铝或氧化铝和焦炭在高温下与氯气作用而制得。为使 用方便,一般制成粉状或小颗粒状。其熔点192℃,180℃开 始升华。无水三氯化铝能溶于大多数的液态氯烷中,并生成 烷基正离子(R+)。也能溶于许多供电子型溶剂中形成络合 物。此类溶剂有SO2、CS2、硝基苯、二氯乙烷等。
因此上述烷基化反应生成的是两者的混合物。 当用碳链更长的卤烷或烯烃与苯进行烷基化时,则 烷基正离子的重排现象更加突出,生成的产物异构体种 类也增多,但支链烷基苯占优的趋势不变。
(3)醇、醛和酮 它们都是较弱的烷化剂,醛、酮 用于合成二芳基或三芳基甲烷衍生物。
醇类和卤烷烷化剂除活性上有差别外,均特别适合 于小吨位的精细化学品,在引入较复杂的烷基时使用。
第二节 烷基化反应的基本原理
2.催化剂
(1)路易斯酸 主要是金属卤化物,其中常用的是AlCl3。 催化活性如下:
AlCl3> FeCl3>SbCl5 >SnCl4> BF3> TiCl4 >ZnCl2 路易斯酸催化剂分子的共同特点是都有一个缺电子中心 原子,如AlCl3分子中的铝原子只有6个外层电子,能够接受 电子形成带负电荷的碱性试剂,同时形成活泼的亲电质点。
C-烷基化反应的可逆性也可由烷基的给电子特性加 以解释。给电子的烷基连于苯环,使芳环上的电子云密 度增加,特别是与烷基相连的那个芳环碳原子上的电子
云密度增加更多,H+进攻此位置较易,转化为σ-络合
物,其可进一步脱除R+而转变为起始反应物。
第二节 烷基化反应的基本原理
(3)烷基正离子能发生重排 C-烷基化中的亲电质 点烷基碳正离子会重排成较稳定的碳正离子。如用正丙 基氯在无水三氯化铝作催化剂与苯反应时,得到的正丙 苯只有30%,而异丙苯却高达70%。这是因为反应过程 中生成的会发生重排形成更加稳定的。
第六重要性
把烃基引入有机化合物分子中的碳、氮、氧等原子 上的反应称为烷基化反应,简称烷基化。所引入的烃基 可以是烷基、烯基、芳基等。其中以引入烷基(如甲基、 乙基、异丙基等)最为重要。广义的烷化还包括引入具 有 各 种 取 代 基 的 烃 基 ( — CH2COOH、—CH2OH、— CH2Cl、—CH2CH2Cl等)。
第二节 烷基化反应的基本原理
此外还有一些其他类型催化剂,如:酸性氧化物, 分子筛,有机铝等。
酸性氧化物,如SiO2-Al2O3也可作为烷基化催化剂。 烷基铝是用烯烃作烷基化剂时的一种催化剂,其中 铝原子也是缺电子的,对于它的催化作用还不十分清楚。 酚铝〔Al(OC6H5)3〕是苯酚邻位烷基化的催化剂,是由 铝屑在苯酚中加热而制得的。苯胺铝〔Al(NHC6H5)3〕 是苯胺邻位烷基化催化剂,是由铝屑在苯胺中加热而制 得的。此外,也可用脂肪族的烷基铝(R3Al)或烷基氯 化铝(AlR2Cl),但其中的烷基必须要与引入的烷基相同。
第二节 烷基化反应的基本原理
以烯烃、醇、醛、和酮为烷化剂时,广泛应用硫酸 作催化剂。在硫酸作催化剂时,必须特别注意选择适宜 的硫酸浓度。因为当硫酸浓度选择不当时,可能会发生 芳烃的磺化,烷化剂的聚合、酯化、脱水和氧化等副反 应。如对于丙烯要用90%以上的硫酸,乙烯要用98%硫 酸,即便如此,这种浓度的硫酸也足以引起苯和烷基苯 的磺化反应,因此苯用乙烯进行乙基化时不能采用硫酸 作催化剂。
第一节 概述
又如,消毒防腐药度米芬(Domiphen Bromide)的 合成,也采用了烷基化反应。
第一节 概述
二、烷基化反应的类型 1.C-烷基化反应 在催化剂作用下向芳环的碳原子上引入烷基,得到取代 烷基芳烃的反应。如烷基苯的制备反应:
2.N-烷基化反应 向氨或胺类(脂肪胺、芳香胺)氨基中的氮原子上 引入烷基,生成烷基取代胺类(伯、仲、叔、季胺)的 反应。如N,N-二甲基苯胺的制备反应:
其以何种形式参加后继反应主要视卤烷结构而定。 由于碳正离子的稳定性顺序是:
+
CH2
~
+
CH2=CH-CH2
+
> R3C
>
+