材料物理化学第四章2
物理化学第四章-化学平衡

平衡 正向自发
ii (产物) ii (反应物) 逆向自发
自发变化的方向:反应总是从化学势较高的一边流向化 学势较低的一边。
1. 化学反应的限度
所有的化学反应既可以正向进行亦可以逆向进行, 且反应正向进行和逆向进行均有一定的程度,反应 物不会完全转化为产物。
典型例子:
H2(g)+I2(g)↔2HI C2H5OH(l)+CH3COOH(l)↔CH3COOC2H5+H2O
任意化学反应: rGm rGm RT ln Qa
rGm ii (产物) jj (反应物)
(化学反应的吉布斯自由能改变量)
rGm
i
i
(产物)
jj (反应物)
(化学反应的标准吉布斯自由能改变量)
1.rGm 是任意反应系统的吉布斯自由能变化
rGm 是任意反应系统的标准摩尔吉布斯自由能变化
rGm 指产物和反应物都处于标准态时,产物的吉布
斯函数与反应物的吉布斯函数总和之差,称为化学
反应的“标准摩尔Gibbs 自由能变化值”,只是温
度的函数。
平衡时Leabharlann gGhHa
A
b
B
r Gm
若上述反应在定温定压下进行,其中各分压是任意的而 不是平衡时的分压,此时反应的吉布斯函数变化为:
非平衡时
rGm gG hH (aA bB )
B
B
RT
ln( PB
/
P )
g[G
RT
ln(
pG
/
p
)]
h[
H
RT
ln(
pH
/
p
)]
a[
A
RT
ln(
物理化学 第4章化学平衡

当 B 0 时 K Kc Kn K y
1/31/2020
祝大家学习愉快,天天进步!
17/114
§4.2-3.有纯态凝聚相参加的理气反应 K
ΔrGm RTln K
J p
B (g)
pB p
(g)
νB(g)
K
B(g)
peq B(g) p
结论:化学反应方程中计量系数呈倍数关系,Δ r Gm
的值也呈倍数关系,而 K 值则呈指数的关系。
1/31/2020
祝大家学习愉快,天天进步!
15/114
§4.2-2.理想气体反应的 K K c K y Kn
气体的组成可用分压力,摩尔分数,浓度表 示,故平衡常数也有不同的表示方法.
K
B
2
(g)
CO
2
(g)
Δ G r m,2 RT lnK 2
(3) CO 2 (g) C(s) 2CO(g) Δ G r m,3 RT lnK3 例 4.5.1
因为: (3) = (1) 2(2)
pB p
B
e
pB yB p
K
B
pB p
B
e
B
yB p
p
B
e
B
y B B
e
B
p
p
B
Ky
p p
B
1/31/2020
祝大家学习愉快,天天进步!
16/114
§4.2—2.理想气体反应的 K K c K y Kn
物理化学:第4章_多组分系统热力学_
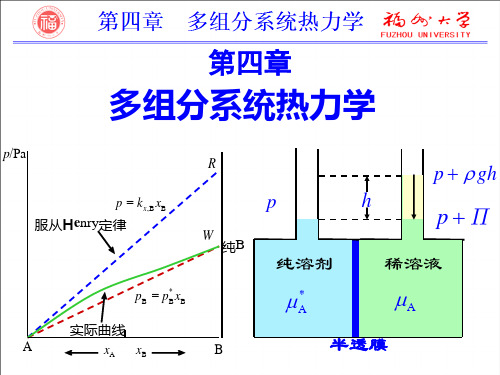
真实混合物:实曲线
Vm xBVB xCVC VB (VC VB)xC
当混合物组成改变时,两组 分偏摩尔体积随之改变,且二者 变化相互关联。
组成接近某纯组分,其偏摩 尔体积也接近该纯组分摩尔体积。
5. 吉布斯 − 杜亥姆方程
对广度量 X (T , p, nB, nC , nD ,) 求全微分:
dX
X T
p,nB
dT
X p
T ,nB
dp
B
X nB
dnB T , p,nC
恒温、恒压
另一方面,由加和公式
,恒温恒压下求导:
比较两式,得
或
或
吉布斯-杜亥姆方程--在一定温度压力下,当混合物
组成变化时,各组分偏摩尔量变化的相互依赖关系。
➢ 系统中各组分的偏摩尔量并非完全独立,而是相 互依存的。
➢ 例:固体溶解、过饱和溶液析出、…
组分B在α、β两相中迁移达平衡的条件:该组分
在两相中的化学势相等。
➢ 物质总是从其化学势高的相向化学势低的相迁移, 直至物质迁移达平衡时为止,此时系统中每个组分在 其所处的相中的化学势相等。
化学势 判据
② 化学平衡
<0:自发不可逆; =0:平衡、可逆
任一化学反应,假定系统已处于相平衡,
任一组分B在每个相中的化学势都相等: Bα B
B
B
整个系统中B组分物质的量的变化量: dnBα dnB
α
BdnB
B
化学平衡时
平衡条件:与化学反应达到平衡的方式无关。
§4.3 气体组分的化学势
1、纯理想气体的化学势 2、理想气体混合物中任一组分的化学势 3、纯真实气体的化学势 4、真实气体混合物中任一组分的化学势
物理化学(王海荣主编)第四章化学平衡概要

上一内容
下一内容
回主目录
返回
2018/10/
用化学反应等温式判断反应方向
化学反应等温式也可表示为:
r Gm RT ln K RT ln Q
Q<Kѳ r Gm <0 反应正向进行; Q =Kѳ r Gm=0 系统处于平衡状态;
Q>Kѳ r Gm >0 反应逆向进行。
NH4HS(s) NH3 (g) H2S(g)
p NH 3 p H 2S 1 p 2 K ( ) 4 p p p
p
上一内容
下一内容
回主目录
返回
2018/10/
复相化学平衡
一定条件下,固体物质分解有气体生成,当达
到平衡时,气相的总压力称作该物质的分解压力。 对复相化学反应来说,分解压力与温度有关,温度 一定时,分解压力一定。温度越高,分解压力越大 ;同一温度下分解压力越大,化合物越不稳定。 当反应系统分解压力等于外压(101.325kPa) 时的温度称为分解温
下一内容
回主目录
返回
2018/10/
标准平衡常数
p B ,eq K (T ) p B
p vB
可以看出: K 1、 p 仅是温度的函数,只随着温度的改变而改变,与物 K 质的浓度或者分压无关。当体系的温度恒定时, p 恒定。 2、由于相对平衡分压 的量纲为一,所以 K p 的量纲也是 一。 3、K p 反映了在一定条件下一个化学反应能够进行的彻底 K 程度。 p 值越大,反应进行的越彻底。一般当 K p >108 ,认为反应进行得完全彻底。
p
vB
上一内容
下一内容
回主目录
物理化学 第四章 多组分系统热力学

Vm
T,p一定
V*m,C VC
V*m,B VB
d c· b·
0 B
a xC
C
图4.1.2 二组分液态混合物的 偏摩尔体积示意图
若B,C形成真实液态混合物: 则混合物体积为由V*m,B至V*m,C的曲线。对于任一 组成a时,两组分的偏摩尔体积可用下法表示: 过组成点a所对应的系统体积点d作Vm-xC曲线的 切线,此切线在左右两纵坐标上的截距即分别 为该组成下两组分的偏摩尔体积VB,VC。
B
系统中各广度量的偏摩尔量: 对于多组分系统中的组分B,有: 偏摩尔体积: VB=(ƽV/ƽnB)T,p,n C 偏摩尔热力学能: UB=(ƽU/ƽnB)T,p,n C 偏摩尔焓: HB=(ƽH/ƽnB)T,p,n C 偏摩尔熵: SB=(ƽS/ƽnB)T,p,n C 偏摩尔亥姆霍兹函数:AB=(ƽA/ƽnB)T,p,n C 偏摩尔吉布斯函数: GB=(ƽG/ƽnB)T,p,n
C
几点说明: (1)偏摩尔量为两个广度性质之比,所以为强度 性质; (2)偏摩尔量的定义中明确是在恒温、恒压及系 统组成不变的条件下,偏导数式的下标为T,p 时才是偏摩尔量; (3)同一物质在相同温度、压力但组成不同的多 组分均相系统中,偏摩尔量不同; (4)若系统为单组分系统,则该组分的偏摩尔量 与该组分的摩尔量相等,即: XB=X*B,m
C
=VB (数学知识:二阶偏导与求导的顺序无关) 得证。
4.2化学势 4.2化学势
1.化学势的定义 混合物(或溶液中)组分B的偏摩尔吉布斯函数GB 定义为B的化学势,用符号μB表示:
μB = GB=(ƽG/ƽnB)T,p,n
def
C
对于纯物质,其化学势等于它的摩尔吉布斯函 数。
第四章 表面与界面

材料的表面与界面
固体(晶体、玻璃体)的表面与内部有什么不同?
实际上晶体和玻璃体:处于物体表面的质点,其 环境和内部是不同的,表面的质点由于受力不均衡而 处于较高的能阶,所以导致材料呈现一系列特殊的性 质。
例如:石英的粉碎。1kg直径为10-2米变成10-9米 ,表面积和表面能增加107倍。
物理性质:熔点、蒸汽压、溶解度、吸附、润湿和烧 结等(微小晶体蒸汽压增大、熔点下降、溶解度增加, 表面上存在着吸附等现象)。
即用于增加物系的表面能。故:∆PdV=γdA
V=4/3πR3 A=4πR2
∴∆P= 2 (球形曲面)
R
对非球形曲面:∆P=
1 r1
1 r2
— 拉普拉斯公式
r1、r2—曲面的主曲率半径
方向:指向曲率中心
2、弯曲表面上的饱和蒸汽压
将一杯液体分散成为微小液滴时,液面就由平面变成凸面, 凸形曲面对液滴所施加的附加压力使液体的化学位增加,从 而使液滴的蒸气压随之增大。所以,液滴的蒸气压必然大于 同温度下平面的蒸气压。它们之间的关系可以用开尔文方程 来描述。
2、固体表面力场
固体内部:质点受到周围质点的控制, 静电平衡、存在力场、力场对称。
固体表面:周期性重复中断,力场对称性破坏, 产生指向空间的剩余力场。
剩余力场表现:固体表面对其它物质有吸引作用 (如润湿、吸附、粘附性)
固体表面上的吸引作用,是固体的表面力场和被吸引质点的力场相 互作用所产生的,这种相互作用力称为固体表面力。
2、浸湿(Soakage)
V S
L
G SL SV
浸湿过程
浸湿过程引起的体系自由能的变化为
G SL SV
如果用浸润功Wi来表示,则是
材料化学第四章固相反应

界I,氧气通过气相传质,于是在相界I上
发生反应:
M 2 g A 3 O l6 e 3 /2 O 2 M 2 O 4 gAl
I
II
MgO MgAl2O4 Al2O3
3/2O2
b
2Al3+
6e
2021/4/30
17
c 表示了3种可能性:
其一为Al3+通过产物而迁移
I
II
到相界I,Mg2+则通过产物 而迁移到相界II,于是在相
2021/4/30
29
然而对于非均相的固相反应,式 (4-9)不能直接用于描述化学反应动 力学关系。因为对于大多数固相反应, 浓度的概念对反应整体已失去了意义。
为了描述非均相的固相反应,人们引 入了转化率G的概念 来取代均相反应中 浓度的概念.
2021/4/30
30
所谓转化率: 参与反应的一种反应物,在反应
MgO与Al2O3要进一步发生反应必须扩散通过反应 产物层。这类反应的进程有多种的可能性,如图4-2 所示。
2021/4/30
13
I
II
MgO MgAl2O4 Al2O3
a
1/2O2
Mg2+
2e
MgO MgAl2O4 Al2O3
3/2O2
b
2Al3+
6e
I
II
MgO MgAl2O4 Al2O3
3Mg2+
氧气在产物层中的扩散系数vd也就是氧气提供的速度41422012112223当整个反应过程达到稳定时氧气提供的速度和氧气消耗的速度相等dxdcdckc由此可见由扩散和化学反应构成的固相反应历程其整体反应速度的倒数为扩散最大速率倒数和化学反应最大速率倒数之为外界氧气浓度43442012112224化学反应速度时化学反应由速度控制表明
物理化学第四章课后答案完整版

第四章多组分系统热力学4.1有溶剂A与溶质B形成一定组成的溶液。
此溶液中B的浓度为c B,质量摩尔浓度为b B,此溶液的密度为。
以M A,M B分别代表溶剂和溶质的摩尔质量,若溶液的组成用B的摩尔分数x B表示时,试导出x B与c B,x B与b B之间的关系。
解:根据各组成表示的定义4.2D-果糖溶于水(A)中形成的某溶液,质量分数,此溶液在20℃时的密度。
求:此溶液中D-果糖的(1)摩尔分数;(2)浓度;(3)质量摩尔浓度。
解:质量分数的定义为4.3在25℃,1 kg水(A)中溶有醋酸(B),当醋酸的质量摩尔浓度b B介于和之间时,溶液的总体积求:(1) 把水(A )和醋酸(B )的偏摩尔体积分别表示成b B 的函数关系。
(2)时水和醋酸的偏摩尔体积。
解:根据定义当时4.4 60℃时甲醇的饱和蒸气压是84.4 kPa ,乙醇的饱和蒸气压是47.0 kPa 。
二者可形成理想液态混合物。
若混合物的组成为二者的质量分数各50 %,求60℃时此混合物的平衡蒸气组成,以摩尔分数表示。
解:甲醇的摩尔分数为58980049465004232500423250....x B =+=4.5 80℃时纯苯的蒸气压为100 kPa ,纯甲苯的蒸气压为38.7 kPa 。
两液体可形成理想液态混合物。
若有苯-甲苯的气-液平衡混合物,80℃时气相中苯的摩尔分数,求液相的组成。
解:4.6在18℃,气体压力101.352 kPa下,1 dm3的水中能溶解O2 0.045 g,能溶解N2 0.02 g。
现将1 dm3被202.65 kPa空气所饱和了的水溶液加热至沸腾,赶出所溶解的O2和N2,并干燥之,求此干燥气体在101.325 kPa,18℃下的体积及其组成。
设空气为理想气体混合物。
其组成体积分数为:,解:显然问题的关键是求出O2和N2的亨利常数。
4.7 20℃下HCl 溶于苯中达平衡,气相中HCl 的分压为101.325 kPa 时,溶液中HCl 的摩尔分数为0.0425。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
与带负电荷的板面形成结合;弱碱
性和中性条件下,棱边局部带正电
荷,易形成边-面结合与边-边结
合。(尤其是高岭石)
2018/12/3 14
(2)、碱金属离子存在以交换原吸附离子: 欲分散,需使粒子间有足够的排斥力
及溶剂化膜
Eiter:
f /K
2
其中f为排斥力;ξ为动电电位; 1/K为扩
散层厚度
2018/12/3 15
当水量多时,水膜的张力松弛下来,粒子间吸引力减弱。 水量少时,不足以形成水膜,塑性也被破坏。
(3). 胶团与介质,胶团与胶团之间的静电引力
可塑性是基于带电粘土胶团与介质中离子之间的 静电引力和胶团间的静电斥力作用的结果。 由于粘土胶团的吸附层和扩散层厚度是随交换性 阳离子的种类而变化的。
当两个颗粒逐渐接近到吸附层以内,斥力开始明 显表现出来,但随着距离拉大,斥力迅速降低。 当引力占优势,它吸引其它粘土粒子包围自己而 呈可塑性。
2018/12/3
(水分子定向排列)
4
水分散
乙醇分散
乙醇+六偏磷酸钠
超细TiO2 ζ=1.2mv
超细TiO2
ζ=-24.8mv
ζ=-63.6mv
Zeta电位表征分散体系稳定性,加入改性剂形成双电 层,双电层斥力降低粒子团聚的引力,实现超微细粒
子均匀分散 Zeta电位的测量表征对粉体, 胶体体系的评价有重要意义
2018/12/3
49
(2)、电解质(吸附或交换阳离子):
由于吸附离子直接影响颗粒间水膜厚度 及吸引力,因而对可塑性影响很大。
吸引力大→克服吸引力位移所需力大→屈服点高
塑性强弱次序同:阳离子交换顺序
此外、提高阳离子交换容量也会改善可塑性
2018/12/3 50
(3)、颗粒细度及形状: 颗粒细 →比表面积大 →毛细管半径小 →成型水分 高→塑性增强; 对不同形状颗粒(板状、短柱状) → 比表面积大 →毛细管力大→塑性增高 。
泥浆的状态可以分为:稀释流
动态、稠化凝聚态以及介于二者之
间的中间触变态
泥浆静 止:凝固
*
扰动: 流动
*
静止:重 新凝固
* 为触变态:似是而非
2018/12/3 21
凝固态――流动态向――凝固态的转
变是逐渐进行的,非突跃性的变化,
并伴随着粘度的变化 触变是介于分散和凝絮之间的中间状态
2018/12/3
2018/12/3 28
C、粘土胶粒大小及形状: 粘土胶粒细小→活性边面多→易形 成触变结构;
粘土胶粒形状不规则→形成卡片 结构时需要的颗粒数少→易形成触 变结构
2018/12/3
29
D、电解质种类及数量:
触变效应与吸附的阳离子及吸附离子的 水化有关。 吸附阳离子→电价低、半径小→粘土的结合 水多→触变性下降
粘土充分胶溶,粘度最低
高ξ电位及厚扩散层的Na黏土具备了胶溶
稳定的条件 (3)、阴离子作用: 阴离子与原粘土上阳离子形
成稳定络合物或不溶物,则碱金属离子的交换反应
更趋完全 完全交换的途径:增加M+浓度,
但易絮凝;选择适合的阴离子
2018/12/3 17
聚合阴离子在胶溶过程中的特殊作用:
硅酸盐、磷酸盐及有机阴离子在水 介质中发生聚合,易牢固地吸附在棱边 上,棱边带正电时起中和作用,棱边带 负电时以物理吸附增加负电荷。结果使 净负电荷增加,增加粒子间排斥力,使 其充分分散
22
2、触变产生的原因――Hoffman模型 Hoffman模型又称为“纸牌结构”
或“卡片结构”
2018/12/3
23
A、泥浆胶溶:粘土颗粒的活性边面
正电荷被中和而使颗粒间相互排斥 而分散
面-面结合
B、不完全胶溶:少量正电荷未被 中和,棱边负电荷不足以排斥板面 负电荷而聚集
结果:形成局部边-面、边-边结合
7
高岭土中加入NaOH
速 度 降
3 x=0.02 mol
4 x=0.2 mol
2 1 x=0 x=0.002 mol 屈服值
流动性
剪切力
H高岭土流变曲线 电解质加入量1→4 ↑
2018/12/3 8
高岭土中加入NaOH
粘度 η
电解质加入量
泥浆稀释曲线
2018/12/3 9
系统粘度随电解质的加入量增加 而出现一个极小值 加入电解质,粘土在介质中分散
2018/12/3 5
1 、无机材料制备中对泥浆的要求(如注 浆成型工艺)
含水量低:孔隙少,收缩率小
流动性好:减小粘度 稳定性高: ζ电位高
2018/12/3
6
在实际的泥浆系统中,通过引 入减水剂(碱性电解质),在降低水量
的同时,提高系统的流动性 (减小粘
度)和稳定性(提高动电电位)
2018/12/3
24
2018/12/3
空隙包裹大 量自由水
六角片状 粒子
局部边- 边(面) 结合
三维网络 架状
高岭石触变结构示意图 疏松网架结构
2018/12/3 25
Hoffman模型:
不完全胶溶的体系中,局部边-面/ 边-边聚合组成三维网络,包裹大量自由水的结 构,使得体系变得粘稠(高粘度);
边-面斥力→稍加剪切力即可破坏三维网 络,释放自由水,体系变得稀释而恢复流 动性; 边-面引力→静止后,重新生成网络结构
影响因素:
1. 电解质种类及加入量,调节稀释剂加入过量或不足:
面-面结合,颗粒紧密,透水性差 部分边-面或边-边结合,颗粒疏松,形成毛细管,透水 性好
2. 泥浆中塑性料与瘠性料配比:塑性料↓,瘠性料↑,透水性 ↑ 3. 颗粒细度:粒径↓,透水性↓
三、粘土的可塑性
1.粘土的可塑性是对泥团系统而言: 粘土+水(一定比例) → 泥团
剪切力>某屈服值
任意塑型
可塑性
2018/12/3
应力消除
形状保持
33
产生可塑性的原因:
一般来说,干的泥料只有弹性,颗粒间表 面力使泥料聚在一起,由于这种力的作用 范围很小,稍有外力即可使泥料开裂。 要使泥料能塑成一定形状而不开裂,则必 须提高颗粒间作用力,同时在产生变形后 能够形成新的接触点, 基于这种认识,有过种种关于泥料塑性的 产生机理。
颗粒间:引力+斥力
含水量大
>20埃 斥力为主 :泥浆状态
颗粒间距远 <20埃 引力增大 含水量低 :塑性体 颗粒接近
不含水, 干泥料
2018/12/3
仅范氏力 :弹性体、
易断裂
46
由于泥团中吸引力主要来自毛细管
力,颗粒间的水膜起到关键的作用。 连续水膜能否维持是决定其塑性的主
要因素
2018/12/3 47
所以:
水量适当 毛细管内: 水膜保持连续性 可塑性
颗粒间距 适当
合力: 吸力为主 毛细管力↓
颗粒间距↑
48
水量太小
水量太大
2018/12/3
水膜中断
水膜太厚
合力:斥力或无吸引力
3、影响泥团可塑性的因素
(1)、矿物组成: 矿物组成不同→比表面积悬殊
→毛细管力相差很大→可塑性不同
蒙脱土>球土>耐火粘土>瓷土
2018/12/3
聚结稳定性的影响因素: 1.双电层斥力 粘土颗粒聚结合并前,必然要移动,随粘 土颗粒一起运动的仅为吸附层的阳离子。 ζ电位表征了实际负电荷的多少。 ζ电位↑→颗粒间斥力越大↑→难以聚结 →聚结稳定性好 2.吸附溶剂化层的稳定作用 吸附→固-液界面表面能→阻止聚结 水化膜→粘度、弹性→妨碍胶粒聚结
因此对应于可塑状态,泥料应有一个最适宜的 含水量,这时它处于松结合水和自由水间的过 渡状态。
(2). 紧薄膜理论
在水存在时,颗粒间隙的毛细管作用对粘土粒子结合的影 响。 认为在塑性泥料的粒子间存在两种力,一是粒子间的吸引 力;另一种是带电胶体微粒间的斥力。
由于在塑性泥粒中颗粒间形成半径很小的毛细管(缝隙), 当水膜仅仅填满粒子间这些细小毛细管时,毛细管力大于 粒子间的斥力,颗粒间形成一层张紧的水膜,泥粒达到最 大塑性。
当斥力大于引力,可塑性较差。因此可以通过阳 离子交换来调节粘土可塑性。
2、粘土中颗粒间作用力
引力 斥力
粘土中颗粒间吸引力:范围为20埃左右
1.范德华力 2.局部边-面的静电引力 3.毛细管力。 其中毛细管力为最主要的作用力
2018/12/3
39
粘土中颗粒间斥力:范围为200埃左右
粘土表面带电荷
(1) 固体键理论
可塑性是由于粘土—水界面键力作用的结果。
粘土和水结合时,第一层水分子是牢固结合的,它不 仅通过氢键与粘土粒子表面结合,同时也彼此联结成 六角网层。
随着水量增加,这种结合力减弱,开始形成较不规则 排列的松结合水层。它起着润滑剂作用,虽然氢键结 合力依然起作用,但泥料开始产生流动性。 当水量继续增加,即出现自由水,泥料向流动状态过 渡。
42
外力较大:颗粒间发生相对位移→ 在 新位置上重新形成吸引力→外力去除
→颗粒在新位置形成平衡(塑性变形)
F
塑性变形
2018/12/3
43
外力很大→颗粒间距离超出吸引力范
围→只有排斥力作用:泥团断裂
F
2018/12/3
泥团断裂
44
吸力为主→可塑
吸引力 水膜厚
含水量 排斥力
2018/12/3
45
在变化中,动力学因素→结构重建需要一定、 时间过程