无参考基因的转录组分析
无参考基因的转录组分析
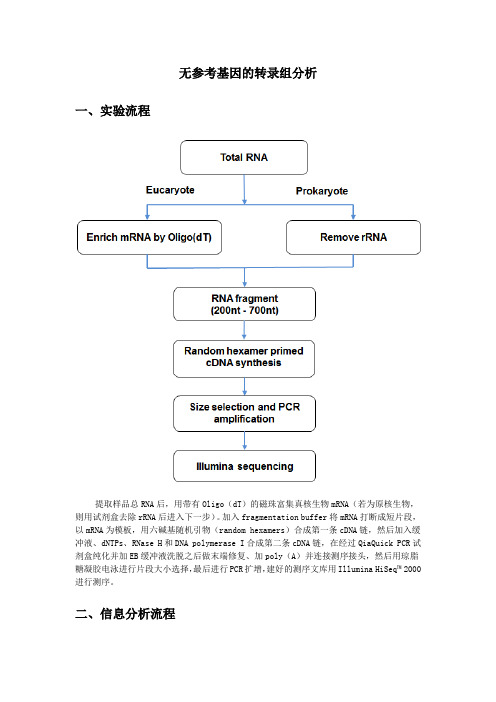
二、信息分析流程
1、产量统计
原始序列数据
测序得到的原始图像数据经 base calling 转化为序列数据,我们称之为 raw data 或 raw reads,结果以 fastq 文件格式存储,fastq 文件为用户得到的最原始文件,里面存储 reads 的序列以及 reads 的测序质量。在 fastq 格式文件中每个 read 由四行描述: \@FC61FL8AAXX:1:17:1012:19200#GCCAAT/1 CCACTGTCATGTGAACATCACAGAGACATTTCTTGA + bbbbbbbbbbbbbbbbbbbbbbbbbaaaaaaaaa_\\
y y
[1]
2 p(i | x) (当 p(i | x) 0.5 时)
i 0 i 0
或者
y y
2(1 p(i | x)) (当 p(i | x) 0.5 时)
i 0 i 0
其中
p(i | x) (
N2 i ) N1
( x i )! N x! y!(1 2 ) ( x i 1) N1
Clean Reads 数据
原始序列数据经过去除杂质后得到的数据。产量统计和后续信息分析分析都基于 Clean Reads。
测序产量统计表格示例
Samples
Total Reads
Total Nucleotides (nt)
Q20 percentage
N percentage
GC percentage *
基因注释到 GO 条目结果文件示例
GO 条目与 All-Unigene 对应结果文件示例
5、Unigene 代谢通路分析
KEGG 是系统分析基因产物在细胞中的代谢途径以及这些基因产物的功能的数据库,利 用 KEGG 可以进一步研究基因在生物学上的复杂行为。 根据 KEGG 注释信息我们能进一步得到 Unigene 的 Pathway 注释。
DNA的质量监测通常有两个方法

2)DNA的质量监测通常有两个方法:首先OD260/OD280比值应该在1.8左右(1.7-1.9),否则意味着DNA样品中存在大量的蛋白质或RNA污染。
其次,琼脂糖电泳分析时应主要以超螺旋条带为主。
最多不超过三条带(分别为超螺旋DNA,线性化DNA和环状DNA)。
否则意味质粒DNA的质量不高,应该重新制备。
2.限制性内切酶的活性1)限制性内切酶一般需要低温保存,而且反复的升降温过程对酶活性的损害很明显。
因而为了确保在有效期内的限制性内切酶不会失活,限制性内切酶的日常保存和使用应当很小。
2)建议购买具有保温功能的冻存盒保存限制性内切酶(-20度),而且取用限制性内切酶时,也应该使用具有保温功能的冻存盒,尽量防止酶的温度反复出现大的波动。
3.限制性内切酶的用量1)限制性内切酶的单位定义通常为:在合适的温度下,完全消化1ugDNA底物所需的酶量定义为一个单位。
2)在这个单位定义中,有几个不确定因素:首先是底物,不同的酶单位定义是选择的底物可能不同(常用的几个底物DNA包括:Lambda DNA ,AD2 DNA 和一些质粒DNA);第二个不确定因素是限制性内切酶在底物DNA上的酶切位点的个数。
由于单位定义中要求完全消化,因而底物上某个酶的酶切位点的个数的多少,就直接影响了该酶的单位定义。
3)因而,在进行酶切时,用1ul酶(一般10IU/ul)消化1ugDNA的通常做法是很不科学的,这也导致在实际工作中,大家要进行多次预实验才能确定最合适酶切条件。
4)以前,我推荐了一个在线的双酶切设计软件,double digestion designer, 可以精确地计算酶切时的限制性内切酶的用量。
使用中,能够注意到,用来进行双酶切的两个酶的用量有时竟然相差近20倍(EcoRI + NheI),而且发现,小片段PCR产物(100-500bp)进行酶切时,需要的酶量比质粒DNA酶切时用量多10倍以上。
5)该软件目前可以免费使用,用户名和密码都是test。
无参考基因的转录组分析

无参考基因的转录组分析无参考基因的转录组分析是指在没有对应基因组序列的情况下,对生物体的转录组数据进行分析,从中获取信息并进行生物学研究。
在无参考基因组的情况下,无法直接对转录组数据进行比对和注释,因此需要采取一些策略和方法来解决这个问题。
1. 转录本组装:通过对转录组数据进行拼接,将转录本组装成单个完整序列,从而获得转录本信息。
这可以使用多个软件来实现,如Trinity、Cufflinks等。
通过对转录本进行定量分析,可以确定各个基因的表达水平。
2. 转录本定量:通过建立转录本的表达矩阵,可以对各个基因的表达水平进行比较和分析。
这可以使用软件如RSEM、eXpress等来完成。
3. 基因功能注释:虽然没有对应基因组序列,但可以利用已知物种的参考基因组信息来进行基因功能注释。
这可以使用一些在线数据库和工具,如Gene Ontology (GO)、KEGG、PANTHER等。
4. 差异表达基因筛选:通过比较不同样本组之间的转录本表达差异,可以筛选出差异表达基因。
这可以使用软件如DESeq2、edgeR等来完成。
5. 寻找新基因:在无参考基因组的情况下,还可以利用转录组数据寻找新基因。
这可以通过比对转录组序列到已知物种的参考基因组上,找出不在参考基因组上的序列,进而预测出新基因。
这可以使用软件如TransDecoder、CPC等来完成。
6.功能富集分析:通过对差异表达基因进行功能富集分析,可以了解这些基因在功能上的特点。
这可以使用一些在线工具和数据库,如DAVID、GSEA等。
7.转录因子分析:转录因子在调控基因的转录过程中起到重要的作用。
通过分析转录因子在转录组中的表达情况,可以了解其在调控过程中的参与情况。
这可以使用一些软件和数据库,如JASPAR、MEME等。
8. 代谢通路分析:通过对差异表达基因进行代谢通路分析,可以了解不同样本组之间在代谢水平上的差异。
这可以使用一些在线工具和数据库,如KEGG、MetaboAnalyst等。
转录组测序以及常用算法简介

转录组测序以及常用算法简介转录组测序,也被称为“全转录组鸟枪法测序”(WTSS),由于转录组测序的高覆盖率,它也被称为深度测序。
它主要利用新一代高通量测序技术,对物种或组织的RNA反转录而成的cDNA文库进行测序,并得到相关的RNA信息。
其研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。
它是指用新一代高通量测序技术,对物种或组织的RNA反转录而成的cDNA文库进行测序,并得到相关的RNA信息。
转录组测序根据有无基因组参考序列分为:有参考基因组的转录组测序,和无参考基因组的de novo测序。
如果有基因组参考序列,可以把转录本映射回基因组,确定转录本位置、剪切情况等更为全面的遗传信息,而这些遗传信息可以广泛应用于生物学研究、医学研究、临床研究中。
虽然转录组测序和基因组测序的步骤大体相同,但是在文库制备和分析方法上却有很大的区别。
在生物信息学领域,序列比对作为识别DNA、RNA和蛋白质相似区域的有效手段,有助于我们更好地研究其结构、功能以及进化方向的关系。
下图简要说明了转录组测序的主要流程:首先将细胞中所有的反转录产物转化为cDNA文库,再将cDNA随机剪切为小DNA片段,并在两端加上接头(Adapter),所得序列通过比对(有参考基因组)或者从头组装de novo(无参考基因组),形成全基因组范围的转录谱。
图1 转录组测序流程图常用算法简介TopHat(/software/tophat/index.shtml)TopHat是Cole Trapnell等人于2009年发表在Bioinformatics上的基于Bowtie的转录组测序比对算法,是马里兰大学生物信息和计算机生物中心,以及加利福尼亚大学伯克利分校数学系和分子细胞生物学系以及哈佛大学的干细胞与再生生物学系联合开发的结果。
它通过超快的高通量短序列比对RNA序列来识别剪切位点。
图2 TopHat流程图TopHat首先先用Bowtie将RNA序列与整个参考基因组进行比对,找到匹配的序列,再用Maq合并匹配的序列,对外显子进行选择性的拼接。
转录组分析学习笔记(持续补充)

转录组分析学习笔记(持续补充)转录组分析流程(有参和⽆参de novo)1. 获得测序数据,Fastq格式,称之为Raw data。
2. 质量检测3. ⽐对Mapping4. Quantification|Quantitation5. 差异表达分析补充:开始项⽬之前,先确⽴合理的⽂件⽬录结构。
【1】Raw Data 处理理论知识⾼通量测序之所以能够能够达到如此⾼的通量的原因就是他把原来⼏⼗M,⼏百M,甚⾄⼏个G的基因组通过物理或化学的⽅式打算成⼏百bp的短序列,然后同时测序。
在测序过程中,机器会对每次读取的结果赋予⼀个值,⽤于表明它有多⼤把握结果是对的。
从理论上都是前⾯质量好,后⾯质量差。
并且在某些GC⽐例⾼的区域,测序质量会⼤幅度降低。
因此,我们在正式的数据分析之前需要对分析结果进⾏质控。
Fastq ⽂件测序给的“原始数据”,称之为Raw Data。
FASTQ是基于⽂本的,保存⽣物序列(通常是核酸序列)和其测序质量信息的标准格式。
其序列以及质量信息都是使⽤⼀个ASCII字符标⽰,最初由Sanger开发,⽬的是将FASTA序列与质量数据放到⼀起,⽬前已经成为⾼通量测序结果的事实标准。
FASTQ⽂件中以四⾏最为⼀个基本单元,并对应⼀条序列的测序信息,各⾏记录信息如下:第⼀⾏记录序列标识以及相关的描述信息,以‘@’开头,为了保证后续分析软件能够区分每条序列,单个序列的标识必须具有唯⼀性;第⼆⾏为碱基序列;第三⾏以‘+’开头,后⾯是序列标⽰符、描述信息,或者什么也不加;第四⾏,是质量信息,长度和第⼆⾏的序列相对应,每⼀个序列都有⼀个质量评分,根据评分体系的不同,每个字符的含义表⽰的数字也不相同。
碱基质量得分与错误率的换算关系: Q = -10log10p(p表⽰测序的错误率,Q表⽰碱基质量分数)ASCII值与碱基质量得分之间的关系:Phred64 Q=ASCII转换后的数值-64Phred33 Q=ASCII转换后的数值-33⽬前illumina使⽤的碱基质量格式为phred+33, 和Sanger的质量基本⼀致(⽼数据建议查看清楚再进⾏后续处理)。
无参转录组 同源基因

无参转录组同源基因
无参转录组分析是指在不预先设定任何参考基因或预设条件下,直接对原始转录数据进行全面的分析,以揭示基因表达的内在规律和特征。
同源基因则是指那些在物种间具有相似或相同功能的基因,它们通常在进化过程中保持保守。
无参转录组同源基因分析的目的是识别和比较不同物种或不同组织之间的同源基因,以了解它们在转录水平上的表达模式和功能。
这种分析有助于发现新的生物标记物、药物靶点或疾病相关基因,并深入了解物种间的进化关系和基因功能。
例如,通过无参转录组分析,科学家可以比较不同组织或不同条件下的转录组数据,以发现与特定生理或病理过程相关的同源基因。
进一步的功能研究可以揭示这些基因在相应过程中的作用,并为其潜在的临床应用提供基础。
总之,无参转录组同源基因分析是一种强大的方法,用于深入了解基因表达的内在规律和特征,以及物种间的进化关系和基因功能。
这种分析有助于发现新的生物标记物、药物靶点或疾病相关基因,并为未来的生物医学研究提供有价值的线索。
无参转录组分析结果的解读

无参转录组分析结果的解读近年来,转录组技术被广泛用于研究基因组功能,而无参转录组(RNA-seq)是用于转录组分析的一种测序技术,其核心原理是通过链特异性加速器(CLASP)进行高通量测序,从而实现对大量基因表达的实时检测,获取转录组的精准分析结果。
无参转录组分析是一个复杂的系统,它结合了基因表达、结构和功能的方面,以及基因之间的分子关系,以便深入的解释和评估基因的表达和调控机制。
无参转录组分析可以协助研究者分析品系和表达模式之间的差异,用于研究基因之间的联系与基因网络,指导药物研发等多方面。
无参转录组分析方法由若干部分组成,主要分为基因表达、基因功能分类和网络功能分析三个步骤。
第一步是基因表达,通过测序产生的数据,采用RPM和FPKM等指标,计算基因表达水平,以定量检测基因在不同样本中的表达变化。
其次是基因功能分类,通过特定算法,检索基因具有特定功能的蛋白质序列,进而确定基因所处细胞环境,了解不同表达基因在特定环境中发挥的作用。
最后是网络功能分析,使用聚类分析,结合元数据构建基因相互作用和调控网络的全貌,以及网络中的细胞因子和调控子的联系,从而可以更加深入的了解基因的表达与功能之间的关系。
无参转录组分析可以提供全面而准确的表达信息,以及基因之间的联系,可以应用于多种领域,如病原学研究、疾病免疫学检测、细胞功能的解析、精准医学建模和药物研发等。
但是,所获得的结果是相对的,必须根据不同的试验要求、受检对象和基因组的不同,以及参考的数据的质量等,综合考虑后才能做出准确的解释和评估。
总之,无参转录组分析技术能够获得准确有效的基因表达分析结果,为生物学研究以及药物研发等提供有力的支持,但也需要对技术、参考数据和评估细节都要进行严谨的评估,才能得到准确的结果。
此外,无参转录组分析的可行性取决于参与者的资源和技术水平,技术突破以及良好的生物组学等应用水平,可以有效提升无参转录组分析的准确性和可靠性,有利于更好的解读分析结果。
利用转录组测序分析大豆矮小突变体中差异表达基因

利用转录组测序分析大豆矮小突变体中差异表达基因近年来,随着测序技术的发展,转录组测序成为研究生物体内基因表达的重要手段之一。
通过转录组测序,可以得知特定生物体在某个生长阶段或环境适应中的基因表达情况。
在大豆研究中,通过转录组测序研究矮小突变体的差异表达基因,可以帮助我们深入了解大豆生长发育中的分子机制。
矮小突变体是指生长期较同个物种的正常个体矮小的一类突变体。
在大豆中,矮小突变体的发现对于提高大豆的产量和抗病能力具有重要意义。
通过转录组测序研究矮小突变体中的差异表达基因,可以揭示矮小突变体生长发育的分子机制,从而为大豆育种提供理论依据。
在利用转录组测序研究大豆矮小突变体中的差异表达基因时,首先需要选择合适的实验材料和对照材料。
一般情况下,可以选择与突变体具有相似生长发育阶段的野生型作为对照材料。
然后,通过高通量测序技术对突变体和对照样品进行测序,获取大量的转录组测序数据。
接下来,对测序数据进行质量控制和过滤,去除低质量的数据和接头序列。
然后使用比对算法将测序数据比对到参考基因组上,得到每个基因的表达情况。
通过比较突变体和对照样品之间的差异表达基因进行筛选,并对其进行功能注释和富集分析。
差异表达基因分析的结果通常可以揭示突变体和对照样品在基因表达水平上的差异,可以给出候选基因的列表。
我们可以进一步对这些候选基因进行生物学实验验证,以了解这些基因在大豆生长发育中的具体功能。
通过转录组测序研究矮小突变体中的差异表达基因,可以明确突变体生长发育过程中哪些基因发生了变化,从而了解突变体的生长发育机制。
此外,通过功能注释和富集分析,我们还可以了解突变体中差异表达基因所参与的生物学过程和通路。
转录组测序研究在大豆育种中的应用前景广阔。
通过研究矮小突变体中差异表达基因,可以为大豆产量和抗病能力的提高提供重要的理论基础。
同时,转录组测序还可以帮助我们发现更多的潜在基因,用于大豆的遗传改良和功能研究。
总之,利用转录组测序研究大豆矮小突变体中的差异表达基因有助于我们了解大豆生长发育过程中的分子机制。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
6、预测编码蛋白框(CDS)
首先,我们按 nr、Swiss-Prot、KEGG 和 COG 的优先级顺序将 Unigene 序列与以上蛋白 库做 blastx 比对(evalue<0.00001),如果某个 Unigene 序列比对上高优先级数据库中的 蛋白,则不进入下一轮比对,否则自动跟下一个库做比对,如此循环直到跟所有蛋白库比对 完。我们取 blast 比对结果中 rank 最高的蛋白确定该 Unigene 的编码区序列,然后根据标 准密码子表将编码区序列翻译成氨基酸序列, 从而得到该 Unigene 编码区的核酸序列 (序列 方向 5'->3')和氨基酸序列。 最后, 跟以上蛋白库皆比对不上的 Unigene 我们用软件 ESTScan (Iseli, Jongeneel et al. 1999)预测其编码区,得到其编码区的核酸序列(序列方向 5'->3')和氨基酸序列。'
[5]
组装出来的序列长度是组装质量的一个评估标准。我们会对组装出来的 Contig、Scaffold、Unigene 做一个长度分布 统计。如下图所示,给出的 bar 图统计 Contig 的长度分布。横坐标是组装出来的 Contig 的长度, 纵坐标是
对
应
长
度
的
Contig
的
数
目
。
组装成功的 Contig 结果在文件夹 1.Contig, Scaffold 相关结果在文件夹 2.Scaffold, Unigene 相关结果在文 件夹 3.Unigene。文件的详细意义可见各个文件夹下面对应的 readme。注:文件夹下面 svg 图可能需要安装 svg 插件才能打开
3、Unigene 功能注释
功能注释信息给出 Unigene 的蛋白功能注释、COG 功能注释。 首先,通过 blastx 将 Unigene 序列比对到蛋白数据库 nr、Swiss-Prot t、KEGG 和 COG (evalue<0.00001) , 得到跟给定 Unigene 具有最高序列相似性的蛋白, 从而得到该 Unigene 的蛋白功能注释信息。 COG 是对基因产物进行直系同源分类的数据库,每个 COG 蛋白都被假定来自祖先蛋白, COG 数据库是基于细菌、藻类、真核生物具有完整基因组的编码蛋白、系统进化关系进行构 建的, 我们将 Unigene 和 COG 数据库进行比对, 预测 Unigene 可能的功能并对其做功能分类 统计,从宏观上认识该物种的基因功能分布特征。
已知, 样本一能比对到所有 Unigene 的总 reads 数为 N1 , 样本二能比对到所有 Unigene 的总 reads 数为 N 2 ,基因 A 在样本一中对应的 reads 数为 x ,在样本二中对应的 reads 数 为 y ,则基因 A 在两样本中表达量相等的概率可由以下公式计算 :Clean Rea Nhomakorabeas 数据
原始序列数据经过去除杂质后得到的数据。产量统计和后续信息分析分析都基于 Clean Reads。
测序产量统计表格示例
Samples
Total Reads
Total Nucleotides (nt)
Q20 percentage
N percentage
GC percentage *
Sample_A
1,634,670
122,600,250
89.47%
0.00%
48.50%
* Total Nucleotides = Total Reads1 x Read1 size + Total Reads2 x Read2 size; Nucleotides are actually clean reads and clean nucleotides;
y y
[1]
2 p(i | x) (当 p(i | x) 0.5 时)
i 0 i 0
或者
y y
2(1 p(i | x)) (当 p(i | x) 0.5 时)
i 0 i 0
其中
p(i | x) (
N2 i ) N1
( x i )! N x! y!(1 2 ) ( x i 1) N1
[1]
H 0 : 某一个基因在两个样本中表达量相同 H a : 某一个基因在两个样本中表达量不同
假设观测到基因 A 对应基因表达量的一小部分,在这种情况下,p(x)的分布服从泊松分布 :
p( x)
e x ( 为基因 A 的真实转录数) x!
然后, 我们对差异检验的 p-value 作多重假设检验校正, 通过控制 FDR (False Discovery Rate)来决定 p value 的域值。假设挑选了 R 个差异表达基因,其中 S 个是真正有差异表达 的基因,另外 V 个是其实没有差异表达的基因,为假阳性结果。希望错误比例 Q=V/R 平均 而言不能超过某个可以容忍的值(比如 1%),则在统计时预先设定 FDR 不能超过 0.01 (Benjamini, Yekutieli. 2001)。 在得到差异检验的 FDR 值同时,我们也根据基因的表达量(RPKM 值)计算该基因在不 同样本间的差异表达倍数。FDR 值越小,差异倍数越大,则表明表达差异越显著。在我们的 分析中,差异表达基因定义为 FDR≤0.001 且倍数差异在 2 倍以上的基因。 得到差异表达基因之后,我们对差异表达基因做 GO 功能分析和 KEGG Pathway 分析。
基因注释到 GO 条目结果文件示例
GO 条目与 All-Unigene 对应结果文件示例
5、Unigene 代谢通路分析
KEGG 是系统分析基因产物在细胞中的代谢途径以及这些基因产物的功能的数据库,利 用 KEGG 可以进一步研究基因在生物学上的复杂行为。 根据 KEGG 注释信息我们能进一步得到 Unigene 的 Pathway 注释。
Unigene 的 COG 功能注释结果样式示例
4、Unigene 的 GO 分类
根据 nr 注释信息我们能得到 GO 功能注释。Gene Ontology(简称 GO)是一个国际标准 化的基因功能分类体系,提供了一套动态更新的标准词汇表(controlled vocabulary)来 全面描述生物体中基因和基因产物的属性。GO 总共有三个 ontology,分别描述基因的分子 功能(molecular function)、所处的细胞位置(cellular component)、参与的生物过程 (biological process) 。 我们根据 nr 注释信息, 使用 Blast2GO 软件 (Conesa, Gotz et al. 2005)得到 Unigene 的 GO 注释信息。Blast2GO 已被其它文献引用超过 150 次,是同行广泛 认可的 GO 注释软件。得到每个 Unigene 的 GO 注释后,我们用 WEGO 软件(Ye, Fang et al. 2006)对所有 Unigene 做 GO 功能分类统计,从宏观上认识该物种的基因功能分布特征。
无参考基因的转录组分析 一、实验流程
提取样品总 RNA 后,用带有 Oligo(dT)的磁珠富集真核生物 mRNA(若为原核生物, 则用试剂盒去除 rRNA 后进入下一步) 。 加入 fragmentation buffer 将 mRNA 打断成短片段, 以 mRNA 为模板,用六碱基随机引物(random hexamers)合成第一条 cDNA 链,然后加入缓 冲液、dNTPs、RNase H 和 DNA polymerase I 合成第二条 cDNA 链,在经过 QiaQuick PCR 试 剂盒纯化并加 EB 缓冲液洗脱之后做末端修复、加 poly(A)并连接测序接头,然后用琼脂 糖凝胶电泳进行片段大小选择00 进行测序。
7、Unigene 表达差异分析
差异表达分析找出在不同样本间存在差异表达的基因, 并对差异表达基因做 GO 功能分 析和 KEGG Pathway 分析。
差异表达基因的筛选
参照 Audic S.等人发表在 Genome Research 上的基于测序的差异基因检测方法 ,我 们开发了严格的算法筛选两样本间的差异表达基因。 筛选差异表达基因所用的到的假设检验的零假设和备择假设如下:
Total Reads and Total
Q20 percentage is proportion of
nucleotides with quality value larger than 20; N percentage is proportion of unknown nucleotides in clean reads; GC percentage is proportion of guanidine and cytosine nucleotides among total nucleotides.
2、组装结果
我们使用短 reads 组装软件 SOAPdenovo 做转录组从头组装。SOAPdenovo 首先将具有 一定长度 overlap 的 reads 连成更长的片段,这些通过 reads overlap 关系得到的不含 N 的组装片段我们称之称为 Contig。 然后, 我们将 reads 比对回 Contig, 通过 paired-end reads 能确定来自同一转录本的不同 Contig 以及这些 Contig 之间的距离,SOAPdenovo 将这些 Contig 连在一起,中间未知序列用 N 表示,这样就得到 Scaffold。进一步利用 paired-end reads 对 Scaffold 做补洞处理,最后得到含 N 最少,两端不能再延长的序列,我们称之为 Unigene。如果同一物种做了多个样品测序,则不同样品组装得到的 Unigene 可通过序列聚 类软件做进一步序列拼接和去冗余处理,得到尽可能长的非冗余 Unigene。 最后,将 Unigene 序列与蛋白数据库 nr、Swiss-Prot、KEGG 和 COG 做 blastx 比对 (evalue<0.00001),取比对结果最好的蛋白确定 Unigene 的序列方向。如果不同库之间的 比对结果有矛盾,则按 nr、Swiss-Prot、KEGG 和 COG 的优先级确定 Unigene 的序列方向, [3] 跟以上四个库皆比不上的 Unigene 我们用软件 ESTScan 预测其编码区并确定序列的方向。 对于能确定序列方向的 Unigene 我们给出其从 5'到 3'方向的序列,对于无法确定序列方向 的 Unigene 我们给出组装软件得到的序列。