前线分子轨道理论HOMO及LUMO的画法
新手友好:高斯09计算DFT中HOMO与LUMO轨道方法

新手入门:Gaussian09通过DFT优化分子结构计算出HOMO/LUMO 轨道基础教程(零基础小白操作指南)所需软件:化学硕士研究生理论计算纯手打经验分享,制作人:坑里的铁蛋菌1.创建打开,File→New→Creat new molecule group在创建面板画出所需计算分子式(以苯环为例)。
图1-1菜单栏图1-2画图界面2.计算菜单栏:依次选择Caculate→Gaussian calculation set up→job type:选择opt+Frep图1-3job type参数Method选择参数如下图:图1-4Method参数其中Basis set中计算方法可根据自身情况选择,图示参数为最简化计算方法。
Method之后的参数系统默认即可,不影响计算结果。
图1-5其它参数界面Submit提交;会提示保存,根据提示界面进行保存(注意保存路径必需全英文);保存文件后弹出转向高斯对话框,点击OK,跳出高斯计算对话框;等待计算结束(计算过程中保持Gauss09Revision计算对话框进行中,Gauss view09可关闭不影响计算)图1-6提交界面图1-7提示保存图1-8保存路径全英文图1-9保存文件后弹出转向高斯对话框图1-10高斯开始计算计算完成,对话框弹出,选择是关闭对话框。
图1-11计算完成弹出对话框3.数据分析打开存储路径,计算完成后,共生成三个文件.图1-12存储路径打开Gauss view09,将后缀为.chk的文件拖入其中。
可以得到经过优化的结构。
图1-13结构优化后的苯环进入菜单栏Edit-->MOs选项,得到窗口如图1-15图1-14MOs选项选中visualize-->单击update,将开始进行电子云渲染。
图1-15MOs点击后呈现页面图1-16电子云渲染4.数据加工渲染结束后,对话框中呈现出HOMO与LUMO轨道的空间电子云分布图,可进行具体分析,单机轨道旁的小方块对HOMO和LUMO轨道进行切换。
光催化 homo lumo位置

光催化 homo lumo位置
Homo(最高占据分子轨道)和Lumo(最低未占据分子轨道)是在光化学中描述分子轨道能量的两个重要概念。
Homo是指电子占据能量最高的分子轨道,而Lumo是指能量最低且未被电子占据的分子轨道。
在光催化中,Homo和Lumo的位置和能量对于反应的进行和催化效果具有重要影响。
以COF-0-3(共价有机框架)为例,通过DFT(密度泛函理论)计算,可以确定COF-0-3的供体-受体配对。
在不同电子状态下,电荷分布表明最高的占据分子轨道(HOMO)分布在整个骨架上,而最低的未占据分子轨道(LUMO)主要分布在苯并噻二唑的电子受体单元上。
这种分布使得轨道在光激发下通过推拉相互作用扩大光吸收范围,促进电荷的有效分离。
对于TPPS/PDI(三亚苯并噻二唑/聚二乙炔)界面,理论计算结果表明,TPPS/PDI界面的LUMO和HOMO分别位于PDI和TPPS上,说明TPPS具有供电子性能,而PDI具有接受电子的性能,结果表明TPPS/PDI界面具有D-A特征。
Chemdraw 绘制分子的homo lomo轨道

ChemBio 3D如何绘制分子轨道作为一款专业的三维分子结构演示软件,ChemBio 3D具有制作结构,立体旋转,读取ChemDraw结构等功能。
而分子轨道理论中的最高占有(HOMO)和最低空轨道(LUMO)在分子反应中也有着重大意义,本实例将以含有双键的最简单分子乙烯来测试双键的反应活性。
1. 基本概念波函数(wave function):在量子力学中,粒子的状态用波函数(满足特定条件的函数)来描述,波函数本身没有明确的物理意义,但波函数的平方描述了粒子在特定区域出现的概率。
波函数能够通过求解薛定谔方程得到,理论上,当确定了一个研究对象的波函数后,就能够获得研究对象的所有性质。
原子轨道(atomic orbitals):原子轨道是指原子中电子的所有可能运动状态,对于单电子原子体系(也就是氢原子),我们能够精确求解薛定谔方程得到一系列正交化的波函数(也就是原子轨道)。
在杂化轨道理论中,原子之间的成键过程被理解为在一定规则下原子轨道的有效重叠,而形成的分子中,电子是被定域在原子周围的。
分子轨道(molecular orbitals):分子轨道是指分子中电子的所有可能运动状态,在分子轨道理论中,分子中的电子被设想为离域在整个分子体系中。
分子轨道波函数通常被表示为组成分子的所有原子的原子轨道的线性组合,能够通过近似求解薛定谔方程得到。
前线轨道(frontier orbitals):前线轨道理论认为,在一个分子的所有分子轨道中,能量最高的占据轨道(HOMO)和能量最低的非占据轨道(LUMO)对分子的反应和性质起着决定性的作用(图1),这些轨道也被统称为前线轨道(也包括SOMO轨道,指的是单电子占据轨道)。
对大多数化学反应而言,在满足分子轨道对称性的条件下,反应在一个反应物的HOMO与另一反应物的LUMO能够产生最大重叠位置及方向上发生。
图1. 分子的HOMO和LUMO轨道2. 生成分子轨道的方法以上的理论表明:对分子轨道具体信息(包括分子轨道能量和分子轨道形状)的了解有助于我们对分子反应性和其他分子性质的了解, 而其中尤其重要的就是分子的HOMO和LUMO轨道。
homo和lumo轨道 计算

homo和lumo轨道计算
HOMO和LUMO是分子轨道理论中的两个重要概念,可以用
来描述分子中电子的能量和分布。
HOMO(Highest Occupied Molecular Orbital)表示最高占据分
子轨道,即能量最低的、被电子填充的分子轨道。
它通常具有较高的电子密度,可以用于描述分子中电子的移动和反应。
LUMO(Lowest Unoccupied Molecular Orbital)表示最低未占
据分子轨道,即能量最高的、未被电子填充的分子轨道。
LUMO通常具有较低的电子密度,可以用于描述分子与其他
物质的化学反应。
针对一个分子,计算HOMO和LUMO可以通过量子化学计算方法来实现,其中最常用的是密度泛函理论(Density Functional Theory,DFT)和分子轨道理论(Molecular Orbital Theory)。
这些计算方法可以通过计算分子的分子轨道能级和
电子密度得到HOMO和LUMO轨道的能量和分布。
要进行HOMO和LUMO的计算,需要使用量子化学计算软件,例如Gaussian、VASP、ORCA等。
入口部分的基础计算步骤
包括构建分子几何结构、选择合适的方法和基组进行计算,并输出能量和分子轨道信息。
根据计算结果,可以得到HOMO
和LUMO的能量和电子分布。
总结起来,计算HOMO和LUMO轨道需要进行量子化学计算,通过相应的软件和方法得出能量和分布信息。
循环伏安曲线怎么算homolumo

循环伏安曲线怎么算homolumo循环伏安曲线是一种常用的电化学分析方法,可以用来研究化合物的电化学性质。
其中,循环伏安曲线中的峰位和峰电位可以提供有关分子的电子结构信息,包括分子的最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)能级。
本文将介绍如何利用循环伏安曲线来计算分子的HOMO和LUMO能级。
我们需要了解循环伏安曲线的基本原理。
循环伏安曲线是通过在电极上施加一定的电势,然后测量电流随时间的变化来得到的。
在循环伏安曲线中,电流随电势的变化呈现出周期性的波动,其中的峰位和峰电位可以提供有关分子的电子结构信息。
接下来,我们需要了解如何从循环伏安曲线中确定分子的HOMO 和LUMO能级。
在循环伏安曲线中,HOMO和LUMO能级分别对应着氧化还原峰的峰位和峰电位。
具体来说,HOMO能级对应着氧化峰的峰位,而LUMO能级对应着还原峰的峰位。
因此,我们可以通过测量循环伏安曲线中的氧化还原峰的峰位和峰电位来确定分子的HOMO和LUMO能级。
我们需要了解如何利用HOMO和LUMO能级来研究分子的电子结构。
HOMO能级代表着分子中最高的占据分子轨道,因此它可以提供有关分子的电子亲和性和化学反应性的信息。
LUMO能级代表着分子中最低的未占据分子轨道,因此它可以提供有关分子的电子亲和性和化学反应性的信息。
通过研究分子的HOMO和LUMO能级,我们可以了解分子的电子结构和化学性质,从而为分子的应用和设计提供有价值的信息。
循环伏安曲线可以用来研究分子的电子结构和化学性质,其中的HOMO和LUMO能级可以提供有关分子的电子结构信息。
通过测量循环伏安曲线中的氧化还原峰的峰位和峰电位,我们可以确定分子的HOMO和LUMO能级,从而了解分子的电子结构和化学性质。
高斯homo和lumo轨道计算

高斯homo和lumo轨道计算(Gaussian HOMO and LUMO Orbital Calculations)一、介绍1. 高斯homo和lumo轨道在量子化学中扮演着重要的角色,它们是分子轨道能级的一种理论描述,对于研究分子的电子结构和化学性质具有重要意义。
2. 本文将探讨高斯homo和lumo轨道计算的原理、方法和应用,旨在帮助读者全面、深入地理解这一主题。
二、原理和方法1. 高斯homo和lumo轨道是通过量子力学计算得出的,其中包括分子轨道理论、量子化学计算方法等。
2. 高斯homo和lumo轨道的计算方法包括密度泛函理论、哈特里-福克方法、从头算方法等,每种方法都有其特定的适用范围和优势。
3. 在计算过程中,需要考虑分子的几何结构、电子态密度、交换相关能等因素,并通过复杂的数学模型和计算工具得出准确的结果。
三、应用和意义1. 高斯homo和lumo轨道的计算结果可以用于解释分子的光学性质、电子亲和性、化学反应活性等化学性质。
2. 通过对高斯homo和lumo轨道的计算与分析,可以帮助科研人员设计新型的药物分子、催化剂和材料,从而推动化学领域的发展。
3. 对高斯homo和lumo轨道的计算结果进行深入研究,还可以揭示分子内部电子结构和化学键性质的微观机制,为理解和预测化学反应提供重要参考。
四、个人观点1. 高斯homo和lumo轨道计算在当今化学研究中具有重要意义,它为我们揭示了分子的电子结构和化学性质提供了强有力的工具。
2. 我个人认为,随着计算方法和计算工具的不断发展,高斯homo 和lumo轨道计算将在未来化学领域继续发挥着重要作用,为新材料、新药物的设计和发现提供有力支持。
五、总结1. 通过本文的深入探讨,相信读者已经对高斯homo和lumo轨道计算有了更全面的了解。
2. 高斯homo和lumo轨道的计算方法和应用意义相当广泛,对于化学研究和应用具有重要价值。
在不同类型的任务中,写手会根据不同的指导进行全面评估,并撰写有价值的文章。
Chemdraw绘制分子的homolomo轨道

Chemdraw绘制分子的homolomo轨道ChemBio 3D如何绘制分子轨道作为一款专业的三维分子结构演示软件,ChemBio 3D具有制作结构,立体旋转,读取ChemDraw结构等功能。
而分子轨道理论中的最高占有(HOMO)和最低空轨道(LUMO)在分子反应中也有着重大意义,本实例将以含有双键的最简单分子乙烯来测试双键的反应活性。
1. 基本概念波函数(wave function):在量子力学中,粒子的状态用波函数(满足特定条件的函数)来描述,波函数本身没有明确的物理意义,但波函数的平方描述了粒子在特定区域出现的概率。
波函数能够通过求解薛定谔方程得到,理论上,当确定了一个研究对象的波函数后,就能够获得研究对象的所有性质。
原子轨道(atomic orbitals):原子轨道是指原子中电子的所有可能运动状态,对于单电子原子体系(也就是氢原子),我们能够精确求解薛定谔方程得到一系列正交化的波函数(也就是原子轨道)。
在杂化轨道理论中,原子之间的成键过程被理解为在一定规则下原子轨道的有效重叠,而形成的分子中,电子是被定域在原子周围的。
分子轨道(molecular orbitals):分子轨道是指分子中电子的所有可能运动状态,在分子轨道理论中,分子中的电子被设想为离域在整个分子体系中。
分子轨道波函数通常被表示为组成分子的所有原子的原子轨道的线性组合,能够通过近似求解薛定谔方程得到。
前线轨道(frontier orbitals):前线轨道理论认为,在一个分子的所有分子轨道中,能量最高的占据轨道(HOMO)和能量最低的非占据轨道(LUMO)对分子的反应和性质起着决定性的作用(图1),这些轨道也被统称为前线轨道(也包括SOMO轨道,指的是单电子占据轨道)。
对大多数化学反应而言,在满足分子轨道对称性的条件下,反应在一个反应物的HOMO与另一反应物的LUMO能够产生最大重叠位置及方向上发生。
图1. 分子的HOMO和LUMO轨道2. 生成分子轨道的方法以上的理论表明:对分子轨道具体信息(包括分子轨道能量和分子轨道形状)的了解有助于我们对分子反应性和其他分子性质的了解, 而其中尤其重要的就是分子的HOMO和LUMO轨道。
ADF教程:如何计算HOMO、LUMO
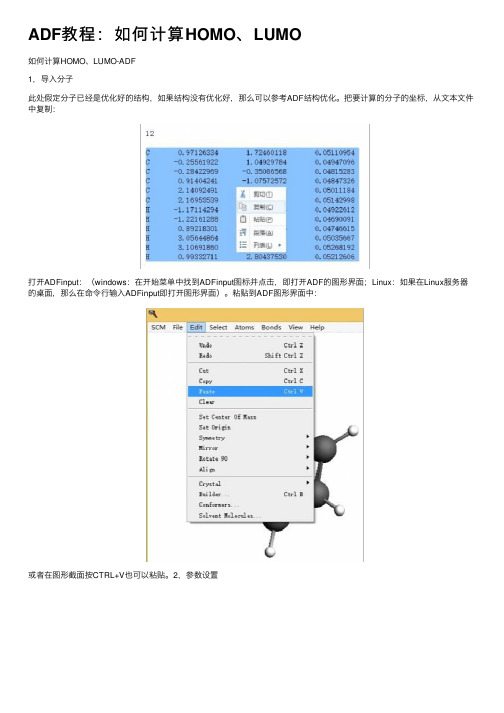
ADF教程:如何计算HOMO、LUMO如何计算HOMO、LUMO-ADF1,导⼊分⼦此处假定分⼦已经是优化好的结构,如果结构没有优化好,那么可以参考ADF结构优化。
把要计算的分⼦的坐标,从⽂本⽂件中复制:打开ADFinput:(windows:在开始菜单中找到ADFinput图标并点击,即打开ADF的图形界⾯;Linux:如果在Linux服务器的桌⾯,那么在命令⾏输⼊ADFinput即打开图形界⾯)。
粘贴到ADF图形界⾯中:或者在图形截⾯按CTRL+V也可以粘贴。
2,参数设置补充说明:◆Total charge指整个体系的带电量,例如本例为中性分⼦,带电量为0;◆Spin polarization指未配对电⼦数,本例中所有电⼦全部配对,因此为0;◆XC potential in SCF指计算使⽤的泛函,本例采⽤B3LYP泛函;◆Basis set指基组,对于⽐较轻的元素,例如CHONS之类,⼏何优化⼀般使⽤DZP基组⾜够,计算性质例如HOMO、LUMO、吸收光谱设为TZP⾜够,如果是较⼤的原⼦例如Au、Pt等,优化时采⽤TZP基组,性质计算时采⽤QZ4P⾜够;◆Frozen core指冻芯近似,⼀定程度上能够节省计算量,但⼀般在结构优化时使⽤,性质计算时不使⽤,因此如本例设置为none,表⽰不使⽤;◆Numerical quality指积分精度,结构优化时normal⾜够,性质计算时good⾜够,如果使⽤metaGGA或者metaHybrid则需要excellent。
3,运⾏计算弹出ADFjobs窗⼝,右边的齿轮表⽰正在运⾏:运⾏完毕后,变成实⼼球:4,查看HOMO、LUMO第⼆列的能级是分⼦的能级,第⼀列,以及后⾯所有列,都是原⼦的能级,这个图反应分⼦轨道与原⼦轨道的关系。
⿏标放到感兴趣的轨道,例如HOMO(最⾼占据轨道),即显⽰轨道的能量和构成,以及占据数:注意,这个能量单位是Hatree,1Hartree=27.2113845eV。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
创作编号:
GB8878185555334563BT9125XW
创作者:凤呜大王*
前线分子轨道理论HOMO及LUMO的画法HOMO:分子轨道中的最高已占轨道,顾名思义,这轨道里面是有电子的。
LUMO:分子轨道中的最低空轨道,这里面没有填充电子,在所有的空轨道中是能量最低的。
如何画出发生周环反应的分子的HOMO以及LUMO呢。
以乙烯为例:
1、要知道乙烯分子中的π电子数:π=2
2、有几个π电子,会组合成相应的轨道数。
故,这里会有两个轨道。
节点分别为1和0;节点数越高能量越大。
第一个轨道,在该轨道中可看到,两个P电子肩并肩重叠可形成π键,两P电子的波相相同。
第二个轨道,在该轨道中,两个P电子波相相反,中间存在节面,不能成键。
第一个轨道,是成键轨道中能量最高的,即我们所谓的HOMO。
第二个轨道,即最低未占轨道,LUMO(需要指出的是,上面虽然画出了两个P电子,但在这里仅仅是用于指出该轨道中的两个电子应该存在的状态如此,并非指此时里面就有两个电子)
如果用电子来表示,就是这样:
在画此类轨道图时,把握住一个原则,即轨道数从1往上,其节点数从0开始增加。
如上图,第一个轨道无节点,第二个轨道有一个节点,如果有第三个轨道,则有2个节点,以此类推。
根据此原则,我们也可以画出1,3-丁二烯的π电子分子轨道位相图,如下:
遵循波粒二象性中的粒子质量守恒原子轨道个数=分子轨道个数
第二个轨道为HOMO,第三个轨道为LUMO
创作编号:
GB8878185555334563BT9125XW
创作者:凤呜大王*。