美国现行GMP(中文版)
美国良好操作规范GMP110

美国良好操作规范(GMP—21CFR Part 110,参考译文)A分部—总则110.3 定义联邦食品、药物及化妆品条例(以下简称条例)第201节中术语的定义和解释适用于本部分的同类术语。
下列定义亦同样适用:1.酸性食品或酸化食品:是指平衡pH值为4.6或低于4.6的食品。
2.适当的:指为达到良好的公共卫生规范的预期目的所需要满足的要求。
3.面糊:是指一种半流体物质,通常包含面粉和其他辅料,食品的主要成分可浸在其中,或用它涂膜,或直接用来形成烘烤的食品。
4.热烫:除坚果和花生外,指在包装前对食品进行足够时间和充分温度的热处理,以使天然形成的酶部分地或完全失活,并使该食品产生物理或生化方面的变化。
5.关键控制点:是指食品加工过程中的一个点,在这个点上控制不当时,就可能造成或导致危害,或使成品受到杂质污染,或造成成品的腐败。
6.食品:指条例201(F)节所定义的食品.包括各种原料和辅料。
7.食品接触面:指与人类食品的接触表面以及在正常加工过程中因污水滴溅污染的并与食品接触的设备和工器具表面。
“食品接触面”包括与食品接触的工器具及设备的表面。
8.批:指在某一段时间生产的由具体代号标记的食品。
9.微生物:是指酵母菌、霉菌、细菌和病毒,包括(但不仅限于)对公众健康有影响的微生物种类。
“有害微生物”这个术语包括对公众健康有影响的,或使食品发酵分解的,或使食品受到杂质污染的、或使食品成为条例所指的劣质食品的微生物。
有时,美国食品与药物管理局在这些法规中使用“微生物”这个词,而不使用包含“微生物”一词的短语。
10.害虫:指令人厌恶的任何动物或昆虫,包括,但不仅限于鸟、啮齿动物、蝇和幼虫。
11.厂房:指用于人类食品加工、包装、标识或存放,或与人类食品的加工、包装、标识或存放有关的建筑物或设施或其中的某些部分。
12.质量控制操作:是指一种有计划的系统操作,并采取一切必要的行动防止食品成为条例所指的劣质食品。
13.返工品:是指由于一些不卫生因素而从加工过程中检选出的、干净的、未被掺杂的食品,或经过重新加工而再次调制,并适于消费的食品。
美国现行GMP(中文版)

美国现行GMP(中文版)美国现行药品生产质量管理规范(cGMP)目录A-总则 (3)B-组织与人员 (3)C-厂房与设施 (4)D-设备 (7)E-成份、药品容器和密封件的控制 (8)F-生产和加工控制 (11)G-包装和标签控制 (13)H-贮存和销售 (16)I-实验室控制 (17)J-记录和报告 (20)K-退回的药品和回收处理 (25)A.总则211·1 范围(a) 本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产质量管理规范(GMP)。
(b) 在本章里的这些针对药品的现行GMP条例和本章600至800的所有部分针对人用生物制品的现行GMP条例,除非明确另有说明者外,应认为是对本部分条例的补充,而不是代替。
本章其他部分或本章600至680各部分和本部分均可适用的条例,前部分的条例可代替本部分条例。
(c) 在考虑经提议的,发表在1978年9月29日联邦注册表(FR)上一项免除时,若产品及其所有成份是以人用物品形式作一般销售和消费,且这些产品根据其预期用途,亦可列入药品的范围内,则不应对这些非处方药(OTC)实施本部分条例,直至进一步的通知为止。
本章110部分和113至119部分的条例用于鉴别这些亦是食品的OTC药品是否按照GMP的要求生产、加工、包装和贮存。
211·3 定义本章210·3的定义适用于本部分。
B. 组织与人员211.22 质量控制部门的职责(a) 本部门有批准和拒收所有成份、药品包装容器、密封件、中间体、包装材料、标签及药品的职责与权力。
复查生产记录的权力,保证不产生差错,或若发生差错,保证他们充分调查这些差错。
本部门负责根据合同,批准或拒收由其它公司,生产、加工、包装或贮存的产品。
(b) 适当的实验室检验设备、批准(或拒收)的各种成份、药品容器、密封件、包装材料及药品,质量控制部门是可以获得的。
(c) 本部门有批准或驳回影响药品的均一性、效价或含量、质量及纯度的所有程序或规格标准的职责。
美国现行GMP规范及实施

ANDA FOR GENERIC PRODUCT 仿制药(非专利药)的ANDA 仿制药(非专利药)的ANDA
《WAXMANHATCH法》,1984年 法 年 允许生产和销售仿制药品, 允许生产和销售仿制药品,即同一种药品可由一家以上 的公司生产。 的公司生产。竞争可以导致药品成本和医疗费用的降低 但要最低限度地影响药品质量。 ,但要最低限度地影响药品质量。仿制品的活性成分和 服用方法必须与拥有注册商标的药品相同。 服用方法必须与拥有注册商标的药品相同。 ANDA要求市场情况介绍,无需临床实验,但要生物 要求市场情况介绍, 要求市场情况介绍 无需临床实验, 等效性报告。 % 等效性报告。80%的非专利药由拥有注册商标的公司来 生产。 生产。
HISTORY OF DRUG REGULATIONS continued
1984 年,《药品价格竞争和专利保护法》 年,《药品价格竞争和专利保护法》 FDA批准 有些ANDA可不进行临床实验 批准, 经FDA批准,有些ANDA可不进行临床实验 许多药品的发明人获得了专利的延期 补偿了FDA 审查过程中的时间损失 补偿了 1992 年,《仿制药实施法案》 年,《仿制药实施法案》 1994 年,《营养增补剂健康和教育法案(DSHEA)》 年,《营养增补剂健康和教育法案(DSHEA)
动物毒理学性和药理学研究 临床协议和研究者资格 化学、 化学、生产和质控 样品和标签 病例报告表和制表 非临床药理学和毒理学 人体药物动力学和生物利用度研究 微生物学 统计学 临床研究
REVIEW PROCESS 审查程序
CDER (药品评审研究中心)接受 药品评审研究中心)接受NDA 中央文件室作出预审查和分类, 中央文件室作出预审查和分类,药品评价 I & II办公室 办公室 预审查可决定申请文件是否符合基本要求 按照用途分类,由专门小组从临床、 按照用途分类,由专门小组从临床、化学和 药理学三方面开始初步审查 与申请人就存在的问题交换意见和要求补充 的资料 作出批准决定
美国药品GMP监管简介课件

美国药品GMP的国际互认
要点一
美国食品药品监督管理局(FDA )与欧盟药品监管机构之间…
FDA与欧盟药品监管机构达成了一项名为“相互承认协定 ”(Mutual Recognition Agreement, MRA)的协议。 根据该协议,欧盟和美国之间互相承认对方的GMP认证, 从而简化了双方市场准入程序。
由FDA制定和发布,规定了药品生产 、控制、包装和标签的最低标准,确 保药品的安全性和有效性。
州级药品GMP法规
各州根据联邦法规制定和实施自己的 药品GMP法规,可增加额外的规定和 标准。
监管流程与方式
现场检查
FDA和州级药品监管部门对药品生产 设施进行定期或不定期的现场检查, 包括生产过程、质量控制、文件记录 等方面。
生产过程要求
操作规范
生产过程应遵循经批准的操作规范,确保产品质量和安全。
工艺控制
生产过程中应对关键工艺参数进行监控和控制,确保产品质 量稳定。
质量控制要求
质量标准
产品应符合预定的质量标准,确保符合相关法规和标准要求。
质量检验
产品应进行严格的质量检验,包括但不限于理化检验、微生物检验和药效学检 验。
加拿大药品GMP
加拿大药品GMP由加拿大卫生部(Health Canada)负责制定和实施。加拿大药品GMP 注重对产品质量的控制,要求企业建立有效的质量控制体系,确保产品符合相关标准和规 定。
日本药品GMP
日本药品GMP由日本医药品医疗器械综合机构(Pharmaceuticals and Medical Devices Agency, PMDA)负责制定和实施。日本药品GMP注重对生产过程的全面控制 ,要求企业建立完善的质量管理体系和生产管理体系。
中国、美国、欧盟GMP中英文版

中华人民共和国卫生部令第 79 号《药品生产质量管理规范(2010年修订)》已于2010年10月19日经卫生部部务会议审议通过,现予以发布,自2011年3月1日起施行。
部长陈竺二○一一年一月十七日第一章总则第一条为规范药品生产质量管理,根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》,制定本规范。
第二条企业应当建立药品质量管理体系。
该体系应当涵盖影响药品质量的所有因素,包括确保药品质量符合预定用途的有组织、有计划的全部活动。
第三条本规范作为质量管理体系的一部分,是药品生产管理和质量控制的基本要求,旨在最大限度地降低药品生产过程中污染、交叉污染以及混淆、差错等风险,确保持续稳定地生产出符合预定用途和注册要求的药品。
第四条企业应当严格执行本规范,坚持诚实守信,禁止任何虚假、欺骗行为。
第二章质量管理第一节原则第五条企业应当建立符合药品质量管理要求的质量目标,将药品注册的有关安全、有效和质量可控的所有要求,系统地贯彻到药品生产、控制及产品放行、贮存、发运的全过程中,确保所生产的药品符合预定用途和注册要求。
第六条企业高层管理人员应当确保实现既定的质量目标,不同层次的人员以及供应商、经销商应当共同参与并承担各自的责任。
第七条企业应当配备足够的、符合要求的人员、厂房、设施和设备,为实现质量目标提供必要的条件。
第二节质量保证第八条质量保证是质量管理体系的一部分。
企业必须建立质量保证系统,同时建立完整的文件体系,以保证系统有效运行。
第九条质量保证系统应当确保:(一)药品的设计与研发体现本规范的要求;(二)生产管理和质量控制活动符合本规范的要求;(三)管理职责明确;(四)采购和使用的原辅料和包装材料正确无误;(五)中间产品得到有效控制;(六)确认、验证的实施;(七)严格按照规程进行生产、检查、检验和复核;(八)每批产品经质量受权人批准后方可放行;(九)在贮存、发运和随后的各种操作过程中有保证药品质量的适当措施;(十)按照自检操作规程,定期检查评估质量保证系统的有效性和适用性。
FDA-GMP中英文对照标准版

DIRECTION OF GMP (GOOD MANUFACTURING PRACTICE )OF RAW MATERIALS BY FDATable of Contents 目录1. INTRODUCTION1.1 Objective 目的1.2 Regulatory Applicability法规的适用性1.3 Scope 范围2. QUALITY MANAGEMENT .质量管理2.1 Principles 总则2.2 Responsibilities of the Quality Unit(s) 质量部门的责任2.3 Responsibility for Production Activities 生产作业的职责2.4 Internal Audits (Self Inspection) 内部审计(自检)2.5 Product Quality Review 产品质量审核3. PERSONNEL 人员3.1 Personnel Qualifications 人员的资质3.2 Personnel Hygiene 人员卫生3.3 Consultants 顾问4. BUILDINGS AND FACILITIES 建筑和设施4.1 Design and Construction 设计和结构4.2 Utilities 公用设施4.3 Water 水4.4 Containment 限制4.5 Lighting 照明4.6 Sewage and Refuse 排污和垃圾4.7 Sanitation and Maintenance 卫生和保养5. PROCESS EQUIPMENT 工艺设备5.1 Design and Construction 设计和结构5.2 Equipment Maintenance and Cleaning 设备保养和清洁5.3 Calibration. 校验5.4 Computerized Systems 计算机控制系统6. DOCUMENTATION AND RECORDS 文件和记录6.1 Documentation System and Specifications 文件系统和质量标准6.2 Equipment cleaning and Use Record 设备的清洁和使用记录6.3 Records of Raw Materials, Intermediates, API Labeling and Packaging Materials原料、中间体、原料药的标签和包装材料的记录6.4 Master Production Instructions (Master Production and Control Records)生产工艺规程(主生产和控制记录)6.5 Batch Production Records (Batch Production and Control Records)批生产记录(批生产和控制记录)6.6 Laboratory Control Records 实验室控制记录6.7 Batch Production Record Review 批生产记录审核7. MATERIALS MANAGEMENT 物料管理7.1 General Controls 控制通则7.2 Receipt and Quarantine 接收和待验7.3 Sampling and Testing of Incoming Production Materials 进厂物料的取样与测试7.4 Storage 储存7.5 Re-evaluation 复验8. PRODUCTION AND IN-PROCESS CONTROLS 生产和过程控制8.1 Production Operations 生产操作8.2 Time Limits 时限8.3 In-process Sampling and Controls 工序取样和控制8.4 Blending Batches of Intermediates or APIs 中间体或原料药的混批8.5 Contamination Control 污染控制9. PACKAGING AND IDENTIFICATION LABELING OF APIs AND INTERMEDIATES原料药和中间体的包装和贴签9.1 General 总则9.2 Packaging Materials 包装材料9.3 Label Issuance and Control 标签发放与控制9.4 Packaging and Labeling Operations 包装和贴签操作10. STORAGE AND DISTRIBUTION.储存和分发10.1 Warehousing Procedures 入库程序10.2 Distribution Procedures 分发程序11. LABORATORY CONTROLS 实验室控制11.1 General Controls 控制通则11.2 Testing of Intermediates and APIs 中间体和原料药的测试11.3 Validation of Analytical Procedures 分析方法的验证11.4 Certificates of Analysis分析报告单11.5 Stability Monitoring of APIs 原料药的稳定性监测11.6 Expiry and Retest Dating 有效期和复验期11.7 Reserve/Retention Samples 留样12. VALIDATION .验证12.1 Validation Policy 验证方针12.2 Validation Documentation 验证文件12.3 Qualification 确认12.4 Approaches to Process Validation 工艺验证的方法12.5 Process Validation Program 工艺验证的程序12.6 Periodic Review of Validated Systems 验证系统的定期审核12.7 Cleaning Validation 清洗验证12.8 Validation of Analytical Methods 分析方法的验证13. CHANGE CONTROL 变更的控制14. REJECTION AND RE-USE OF MATERIALS.拒收和物料的再利用14.1 Rejection 拒收14.2 Reprocessing 返工14.3 Reworking 重新加工14.4 Recovery of Materials and Solvents 物料与溶剂的回收14.5 Returns 退货15. COMPLAINTS AND RECALLS 投诉与召回16. CONTRACT MANUFACTURERS (INCLUDING LABORATORIES)协议生产商(包括实验室)17. AGENTS, BROKERS, TRADERS, DISTRIBUTORS, REPACKERS, AND RELABELLERS 代理商、经纪人、贸易商、经销商、重新包装者和重新贴签者17.1 Applicability 适用性17.2 Traceability of Distributed APIs and Intermediates已分发的原料药和中间体的可追溯性17.3 Quality Management 质量管理17.4 Repackaging, Relabeling, and Holding of APIs and Intermediates原料药和中间体的重新包装、重新贴签和待检17.5 Stability 稳定性17.6 Transfer of Information 信息的传达17.7 Handling of Complaints and Recalls 投诉和召回的处理17.8 Handling of Returns 退货的处理18. Specific Guidance for APIs Manufactured by Cell Culture/Fermentation用细胞繁殖/发酵生产的原料药的特殊指南18.1 General 总则18.2 Cell Bank Maintenance and Record Keeping 细胞库的维护和记录的保存18.3 Cell Culture/Fermentation 细胞繁殖/发酵18.4 Harvesting, Isolation and Purification 收取、分离和精制18.5 Viral Removal/Inactivation steps 病毒的去除/灭活步骤19. APIs for Use in Clinical Trials 用于临床研究的原料药19.1 General 总则19.2 Quality 质量19.3 Equipment and Facilities设备和设施19.4 Control of Raw Materials 原料的控制19.5 Production 生产19.6 Validation 验证19.7 Changes 变更19.8 Laboratory Controls 实验室控制19.9 Documentation 文件20. Glossary 术语1. INTRODUCTION 1. 简介1.1 Objective 1.1目的This document is intended to provide guidance regarding good manufacturing practice (GMP) for the manufacturing of active pharmaceutical ingredients (APIs) under an appropriate system for managing quality. It is also intended to help ensure that APIs meet the quality and purity characteristics that they purport, or are represented, to possess.本文件旨在为在合适的质量管理体系下制造活性药用成分(以下称原料药)提供有关优良药品生产管理规范(GMP)提供指南。
美国cGMP-中英文对照
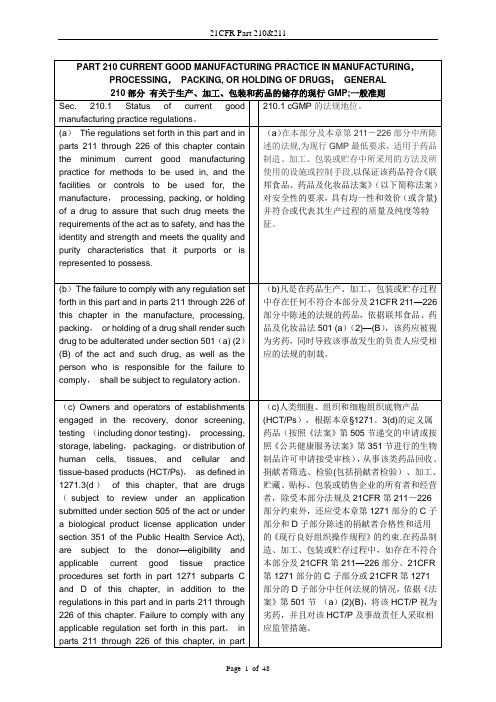
PART 210 CURRENT GOOD MANUFACTURING PRACTICE IN MANUFACTURING,PROCESSING,PACKING, OR HOLDING OF DRUGS;GENERAL210部分有关于生产、加工、包装和药品的储存的现行GMP;一般准则Sec. 210.1 Status of current goodmanufacturing practice regulations。
210.1 cGMP的法规地位。
(a)The regulations set forth in this part and in parts 211 through 226 of this chapter contain the minimum current good manufacturing practice for methods to be used in, and the facilities or controls to be used for, the manufacture,processing, packing, or holding of a drug to assure that such drug meets the requirements of the act as to safety, and has the identity and strength and meets the quality and purity characteristics that it purports or is represented to possess. (a)在本部分及本章第211-226部分中所陈述的法规,为现行GMP最低要求,适用于药品制造、加工、包装或贮存中所采用的方法及所使用的设施或控制手段,以保证该药品符合《联邦食品、药品及化妆品法案》(以下简称法案)对安全性的要求,具有均一性和效价(或含量)并符合或代表其生产过程的质量及纯度等特征。
最新的美国化妆品良好生产规范指南(GMP)

最新的美国化妆品良好生产规范指南(GMP)The Federal Food, Drug and Cosmetic Act1 prohibits the introduction or delivery for introduction into interstate commerce of cosmetics that are adulterated or misbranded (Sec. 301).联邦食品,药品和化妆品法案(The Federal Food, Drug and Cosmetic Act, 以下简称FD&C 法案)禁止在州际直接贸易的化妆品是掺杂的或贴假标签的情况。
(Sec. 301)A cosmetic may be deemed adulterated (Sec. 601) for essentially four reasons, namely:以下 4 种情况下,化妆品被认为是可能掺杂的。
(Sec. 601)1.It may be injurious to users under conditions of customary use because it contains, or its container iscomposed of, a potentially harmful substance.客户在使用过程中,由于化妆品本身含有或在包装容器中有潜在的,对人体有害的成份而使用户受到伤害的。
2.It contains filth.本身含有不洁成份的。
3.It contains a non-permitted, or in some instances non-certified, color additive.本身含有禁用成份,例如:未认可的色素添加剂。
4.It is manufactured or held under insanitary conditions whereby it may have become injurious to usersor contaminated with filth.在不卫生条件下生产的,或保留的,可导致产品伤害用户有害或被不洁成份所污染。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
美国现行药品生产质量管理规范(cGMP)目 录A- 总则………………………………………………………………3B- 组织与人员 (3)C- 厂房与设施 (4)D- 设备………………………………………………………………7E- 成份、药品容器和密封件的控制 (8)F- 生产和加工控制 (11)G- 包装和标签控制 (13)H- 贮存和销售 (16)I- 实验室控制 (17)J- 记录和报告 (20)K-退回的药品和回收处理 (25)A.总 则211·1 范围(a) 本部分的条例包含人用或兽用药品制备的现行最低限度的药品生产质量管理规范(GMP)。
(b) 在本章里的这些针对药品的现行GMP条例和本章600至800的所有部分针对人用生物制品的现行GMP条例,除非明确另有说明者外,应认为是对本部分条例的补充,而不是代替。
本章其他部分或本章600至680各部分和本部分均可适用的条例,前部分的条例可代替本部分条例。
(c) 在考虑经提议的,发表在1978年9月29日联邦注册表(FR)上一项免除时,若产品及其所有成份是以人用物品形式作一般销售和消费,且这些产品根据其预期用途,亦可列入药品的范围内,则不应对这些非处方药(OTC)实施本部分条例,直至进一步的通知为止。
本章110部分和113至119部分的条例用于鉴别这些亦是食品的OTC药品是否按照GMP 的要求生产、加工、包装和贮存。
211·3 定义本章210·3的定义适用于本部分。
B. 组织与人员211.22 质量控制部门的职责(a) 本部门有批准和拒收所有成份、药品包装容器、密封件、中间体、包装材料、标签及药品的职责与权力。
复查生产记录的权力,保证不产生差错,或若发生差错,保证他们充分调查这些差错。
本部门负责根据合同,批准或拒收由其它公司,生产、加工、包装或贮存的产品。
(b) 适当的实验室检验设备、批准(或拒收)的各种成份、药品容器、密封件、包装材料及药品,质量控制部门是可以获得的。
(c) 本部门有批准或驳回影响药品的均一性、效价或含量、质量及纯度的所有程序或规格标准的职责。
(d) 适用于本部门的职责与程序,应成文字材料,并应遵循。
211.25 人员资格(a) 每位从事药品生产、加工,包装或仓贮工作的人员,应接受培训、教育及有实践经验,完成委派的各项职务。
培训是按照现行GMP(包括本章中的现行GMP条例和这些条例要求的成文程序)中涉及雇员的内容。
邀请合格人员指导,并连续多次培训,保证雇员熟悉现行GMP(CGMP)对他们的要求。
(b) 负责监督药品的生产、加工、包装或仓贮工作的每一个人员,应受教育、培训及有经验,完成委派的各项职务。
以此作为提供药品具有安全性、均一性、效价或含量、质量及纯度的保证。
(c) 有足够量执行和监督每种药品的生产、加工、包装或仓贮的合格人员。
211·28人员职责(a) 从事药品生产、加工、包装或仓贮的人员,应穿着适合于其履行职责的清洁衣服。
按需要,头部、脸部、手部、臂部加外罩,防止药物受污染。
(b) 人员保持良好的个人卫生和健康。
(c) 未经监督人员允许,其他人不能进入限制进去的建筑物和设施。
(d) 任何人,在任何时间,明显地表现出带有影响药品安全性和质量的疾病或开放性损伤,应避免接触各种成份、药品容器、包装设备、密封件、中间体,直至病愈或经医学测定认为对药品安全性及质量无危害性时为止。
教育所有人员,报告监督人员对药品有不利影响的健康情况。
211·34顾问为了对问题提出意见,聘请顾问。
顾问应对药品生产、加工、包装或仓贮提出建议,他们受过足够的教育,培训,且有丰富的实践经验。
保留他们的姓名、地址、任何的顾问资格及服务形式等履历资料。
C. 厂房和设施211·42设计与建造特征(a) 任何用于某类药品生产、加工、包装或贮存的厂房或建筑群,大小适宜,结构与位置使其易于清洁、保养、适合操作。
(b) 建筑物有足够空间来有条理地安装设备和放置材料,避免不同类的成份、药品容器、密封件、标签、中间体或药品等相互混放,防止污染。
通过厂房的上述物料其流向在设计时要防止污染。
(c) 操作应在明确规定的,大小适小的地区内进行。
这些地区按规定各自分隔开,以防污染。
下列操作须在单独的地区内进行:(1) 发放给生产或包装前,质量控制部门取样期间,成份、药品容器、密封件及标签的签收、鉴别、贮存及拒收。
(2) 在处理前,拒收的成份,药品容器、密封件及标签的贮存。
(3) 已发放的成份、药品容器、密封件及标签的贮存。
(4) 中间体的贮存。
(5) 生产与加工操作。
(6) 包装和贴标签操作。
(7) 药品发放前的隔离贮存。
(8) 发放后药品的贮存。
(9) 控制与实验室操作。
(10) 无菌操作及有关要求:(I) 地板、墙壁和天花板平滑坚硬、表面易清洁;(Ⅱ) 温度与湿度控制;(Ⅲ) 空气经高效过滤器,在正压下过滤,层流或非层流均可;(Ⅳ) 环境监测系统;(V) 创造无菌环境,房间和设备的清洁、消毒系统;(Ⅵ) 探制无菌环境的设备维修系统。
(d) 青霉素生产、加工及包装设备与生产其他人用药品的设备分开。
211·44照明所有地区均须充足的照明。
211·46通风、空气过滤、空气加热与冷却(a) 提供足够的通风。
(b) 提供足能控制空气正压、微生物、尘土、湿度和温度的设备,适应药品生产、加工和贮存之需要。
(c) 空气过滤系统,包括预过滤器和微粒物质空气过滤器。
空气经过滤才送至生产区,如果空气是再循环到生产区,应测量尘埃含量,控制从生产区带来之尘埃。
在生产区,生产中发生空气污染,应以排气系统或其他系统充分抽出空气,控制污染。
(d) 青霉素生产、加工和包装的空气输送系统应与其他人用药品的空气输送系统完全分开。
211·48管件(a) 在持续正压下,应对药品无污染的管道系统内供应饮用水。
饮用水应符合环境保护机构制订的“基本饮用水条例”标准(40 CFR l4l 部分)。
不符合该标准的水,不许进入水系统。
(b) 排水设备应有足够的大小,可直接连接排水管及安装防止虹吸倒流的空气破坏设备或其他机械设备。
[43 FR 45077,1978年9月29日,修正于48 FR 11426,1983年3月18日]。
211·50污水和废料来自厂房和附近建筑物的污水、垃圾及其他废料,用安全、卫生的方法处理。
211·52洗涤和盥洗设备提供适当的洗涤设备,包括热、冷水、肥皂、清洁剂、空气干燥器或专用毛巾及易进入厕所的清洁设备。
211·56环境卫生(a) 所有用作药品生产、加工、包装及贮存的厂房应保持清洁、卫生的环境,且不受啮齿动物、鸟类、昆虫及其他害虫侵扰(实验动物除外)。
垃圾和有机废料,定时以卫生的方法控制与处理。
(b) 填写分配卫生清洁任务和详细的清洁项目、方法、设备、用于清洁厂房和设施的材料的—览表。
(c) 填写适用的杀鼠剂、杀昆虫剂、杀真菌剂、熏蒸剂、去垢剂和消毒剂一览表。
防止这些物品对设备、成分、药品容器、密封件、包装材料、标签或药品污染。
除依据联邦杀虫剂、杀真菌剂及杀鼠剂法规(7U.S.C135)已登记和使用的品种外,其他的不用。
(d) 在平常的操作过程中,环境卫生工作应适于合同工或临时工及专职人员去完成。
21l·58保养任何用于药品生产、加工、包装或贮存的厂房应保持保养良好状态。
D.设 备211·63设备的设计、尺寸及位置药品生产、加工、包装或贮存设备,设计合理,大小适当,布置合理,便于操作、清洁和保养。
211·65设备制造(a) 设备表面与各种成分、中间体或药品接触,不产生化学反应和吸附作用。
保证药品的安全性、均一性、效价或含量、质量或纯度不变。
(b) 操作所需之物质,如润滑剂、冷却剂等不能进入设备里,与成分,药品容器、封口物品、中间体或药品接触,保证药品的安全性、均一性、效价或含量、质量或纯度不变。
211·67设备清洁与保养(a) 相隔一定时间,对设备与工具进行清洁、保养和消毒,防止出故障与污染,影响药品的安全性、均一性、效价或含量、质量或纯度。
(b) 制订药品生产、加工、包装或贮存设备(包括用具)的清洁和保养文字程序,并执行。
这些程序包括,但不一定限于以下内容:(1) 分配清洁,保养任务。
(2) 保养和清洁细目一览表,包括消毒一览表。
(3) 详细说明用于清洁和保养的设备、物品和方法。
拆卸和装配设备的方法必须保证适合清洁和保养的要求。
(4) 除去或擦去前批遗留物的鉴定。
(5) 已清除了污染的清洁设备的保护。
(6) 使用前检查清洁的设备。
(c) 保留保养、清洁、消毒的记录。
按2111·80及211·182的说明检查。
211·68自动化设备、机械化设备和电子设备(a) 用于药品生产、加工、包装和贮存的自动化、机械化或电子设备,包括计算机或其他类型的设备。
按惯例,对其设计之成文条款作标定、检查或核对,保证其工作性能良好。
保留检查、标定、核对等文字记录。
(b) 对保障重要生产变化的计算机或有关系统进行操作培训。
操作记录或其他记录只能由被认可的人员制订。
向计算机或有关系统输入或从中输出的各种方案、其他记录或资料,应核查其准确性。
输入计算机或有关系统内的档案资料,除与实验室共同分析计算的结果可消除外,其他的应保留。
文字记录与相应的证明资料一起保存。
事先设计好的硬件复制品或多种选择系统,如复印件、磁带或微型胶卷等,保证其支持资料正确、可靠及完整。
出现资料改动、非人为消除或遗失时,应维修。
211.72过滤器用于生产、加工的液体过滤器或人用注射药品的包装材料不许释放出纤维物进入产品。
除必不得以,不在生产、加工中使用释放纤维物的过滤器或注射药品的包装材料。
若必须使用一种能释放纤维素的过滤器,最后应使用—非释放纤维物、平均最大孔径为0.22微米(如实际生产条件限制,可用0.45微米)的附加过滤器过滤,降低注射剂内微粒量。
使用含石棉的过滤器,最后用或不用特殊非释放纤维过滤器均可以,但要根据FDA有关部门提供的,关于该非释放纤维过滤器会或可能损害注射剂的安全性和有效性的证据而定。
E.成分、药品容器和密封件的控制211·80总要求(a) 有文字详细说明成份、药品容器、密封件的签收、鉴定、贮存、装运、取样、检验和批准或拒收等程序,并遵循。
(b) 成份、药品容器和密封件应专人管理和在防止污染的环境下贮存。
(c) 药品容器的包装袋或包装箱或密封件应离地面放置,保持适当间隔,便于清洁和检查。
(d) 用明显的已接收的每装货量中的批号代码对成份、药品容器或密封什加以鉴别。
此代码用来记录每批货的放置地方。