可疑非预期严重不良反应SUSAR的个例报告3页
严重不良事件报告表
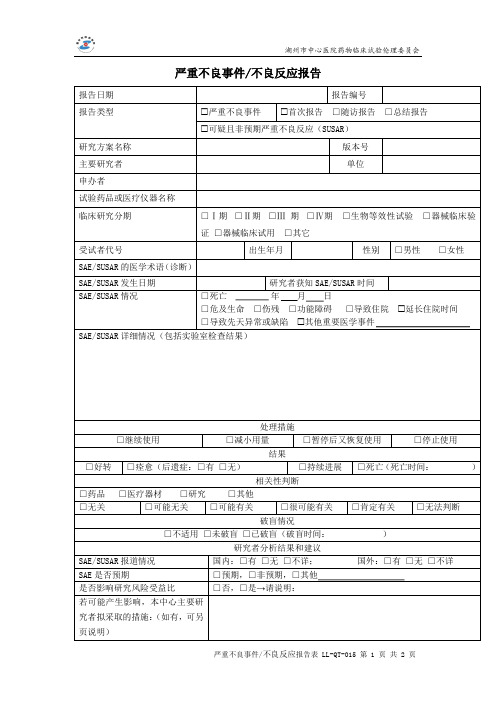
延长住院时间
□继续使用
处理措施
□减小用量
□暂停后又恢复使用
□停止使用
结果
□好转 □痊愈(后遗症:□有 □无)
□持续进展 □死亡(死亡时间:
Байду номын сангаас
)
□药品 □医疗器材 □研究
相关性判断 □其他
□无关
□可能无关
□可能有关
□很可能有关
破盲情况
□肯定有关
□无法判断
□不适用 □未破盲 □已破盲(破盲时间:
)
研究者分析结果和建议
证 □器械临床试用 □其它
受试者代号
出生年月
性别 □男性 □女性
SAE/SUSAR 的医学术语(诊断)
SAE/SUSAR 发生日期
研究者获知 SAE/SUSAR 时间
SAE/SUSAR 情况
□死亡
年月日
□危及生命 □伤残 □功能障碍 □导致住院
□导致先天异常或缺陷 其他重要医学事件
SAE/SUSAR 详细情况(包括实验室检查结果)
湖州市中心医院药物临床试验伦理委员会
严重不良事件/不良反应报告
报告日期
报告编号
报告类型
严重不良事件 首次报告 □随访报告 □总结报告
可疑且非预期严重不良反应(SUSAR)
研究方案名称
版本号
主要研究者
单位
申办者
试验药品或医疗仪器名称
临床研究分期
□Ⅰ期 □Ⅱ期 □Ⅲ 期 □Ⅳ期 □生物等效性试验 □器械临床验
是否需要修改研究方案? 是否需要修改知情同意书?
研究者签名
湖州市中心医院药物临床试验伦理委员会
是 □是
□否 □否
□计划更新 □计划更新
不良事件及严重不良事件处理和报告管理制度

不良事件及严重不良事件处理和报告管理制度不良事件及严重不良事件处理和报告管理制度机密文件未经许可不得擅自使用、泄露、公布及出版1.目的:建立本机构药物临床试验不良事件和严重不良事件处理和报告的管理制度,保障受试者的合法权益和生命安全。
2.适用范围:所有本临床试验机构进行的所有临床试验的不良事件及严重不良事件处理和报告过程。
3.责任人:机构项目负责人及专业负责人4.依据:《中华人民共和国药品管理法》《中华人民共和国药品管理法实施条例》《药物临床试验质量管理规范》(2003版)《药品不良反应报告和监测管理办法》5.定义:不良事件(Adverse Event),病人或临床试验受试者接受一种药物后出现的不良医学事件,但并不一定与治疗有因果关系。
严重不良事件(Serious Adverse Event),临床试验过程中发生需住院治疗、延长住院时间、伤残、影响工作能力、危及生命或死亡、导致先天畸形等事件。
非预期药物不良反应(Unexpected Adverse Drug Reaction,,U-ADR):是性质或严重程度与相应的试验药物资料不一致的药物不良反应。
6.内容:6.1不良事件6.1.1明确不良事件的定义和范围:6.1.1.1不良事件(AE)可以与使用(研究)药物在时间上相关的任何不利的和非预期的体征(包括异常的实验室发现)、症状或疾病,而不管其是否与药物有关;6.1.1.2实验室检查结果异常具有临床意义的定义:受试者实验室检查结果试验前正常,试验后如果数值超过正常值高限或下限150%;或试验前异常,试验后数值比试验前的异常值增高或降低200%。
或研究人员认为有临床意义者可不受此限制。
“实验室检查结果如有异常且具临床意义”的事件按不良事件的5级评定标准判断与试验药物的关系。
6.1.1.3研究药物是指在研究中各阶段所服用的待评估药物,包括对照药品和相应的安慰剂;6.1.1.4在申办者提供的并由伦理委员会批准的临床试验方案中会对不良事件的例外排除情况有所界定,该类事件不作为不良事件处理。
临床试验非预期严重不良事件审查SOP

临床试验非预期严重不良事件审查SOP临床试验非预期严重不良事件审查SOPL目的:为使伦理委员会非预期严重不良事件审查的受理、处理、审查、传达决定、文件存档的工作有章可循,特制定本规程,以从程序上保证非预期严重不良事件审査工作的质量。
2.可疑且非预期严重不良反应(SUSAR),指临床表现的性质和严重程度超出了试验药物研究者手册、已上市药品的说明书或者产品特性摘要等已有资料信息的可疑并且非预期的严重不良反应。
研发期间安全性更新报告(DSUR),是申办者对报告周期内收集到的与在研药物(无论上市与否)相关的安全性信息进行全面深入的年度回顾和评估。
本SOP适用于伦理委员会对非预期严重不良事件报告所进行的审查。
方案定义的需要向伦理委员会报告的非预期严垂不良事件的频率或严重性非预期性地增加,以及非预期、与研充相关、且给受试者或他人带来风险的不良事件,参照本规程执行。
申办者汇总的多中心临床试验的安全性信息报告,主要研究者需阅读并签字后向伦理委员会报告,参照本规程执行。
3.职责3.1伦理委员会秘书:受理并处理送审材料,为委员审査工作提供服务:传达决定:文件存档。
3.2主审委员:会前审査主审项I」的送审文件,填写审査工作表;会议审査作为主要发言者,提问和发表审査意见。
3.3独立顾问:会前审査咨询项目的送审文件,填写咨询工作表;受邀参加审查会议,陈述意见。
3.4委员:会前对审査项目进行预审;参加审査会议,审査毎一项目,提问和发表审查意见,以投票方式做出审查决定。
3.5主任/副主任委员:主持审查会议,审签会议记录,审核、签发审査决定文件。
4.流程图:5 .流程的操作细则5.1受理形式审查1送审文件的完整性①1E预期严重不应事件审査的送审文件包括:非预期严重不良事件报告,以及非预期严重不艮事件的后续报告。
②其他中心发生的非预期的药物严重不良反应,送审文件需包括该中心的伦理审査意见(如有)。
③多中心临床试验的安全性信息报告。
药品不良反应评价和上报

立即
新的、严重的
15日
一般的
30日
国家报告时限要求
奖励和惩罚
奖励制度:对上报药品不良反应/事件报告数量较多、质量较高的科室和个人,医院年终给予通报表彰和不同等级的奖励。 各临床科室药品不良反应/事件报告情况纳入综合医疗质量管理范围,对经医院药品不良反应监测小组查实后瞒报或漏报的,扣科室综合质量考评分;未采取有效措施控制严重药品不良反应重复发生并造成严重后果的,依照有关规定给予行政处分,医院将追究其责任,并根据情况做出相应处罚。
2
潜伏期较长 无明确时间关系 难预测!!
3
”
关联性评价—药品不良反应判断
用药与不良反应/事件的出现有无合理的时间关系?
ADR分析,主要遵循以下五条原则
反应是否符合该药已知的不良反应类型? 停药或减量后,反应是否消失或减轻? 再次使用可疑药品是否再次出现同样反应/事件? 反应/事件是否可用并用药的作用、患者病情的进展、其他治疗的影响来解释?
《药品不良反应/事件报告表》填报要求
《药品不良反应/事件报告表》的填报内容应 真实、完整、准确、及时 报表填写不规范,不完整将影响ADR报告表数据的统计、分析和使用。
药品不良反应报告注意事项
纸质报表填写
A
在线网络填报
B
药品不良反应报告注意事项
登录页面
药品不良反应报告填写注意事项
首页
药品不良反应填报要求
新的药品不良反应 指药品说明书中未载明的不良反应。说明书中已有描述,但不良反应发生的性质、程度、后果或者频率与说明书描述不一致或者更严重的,按照新的药品不良反应处理 。
药品不良反应的定义
可疑的、非预期的严重不良反应(SUSAR Suspected Unexpected Serious Adverse Reaction) 指致死或危及生命的可疑且非预期的严重不良反应。 何谓“非预期”:指不良事件的性质、严重程度或频率超出现有的临床试验资料信息: 未上市药物的研究者手册、药品说明书、试验方案、IB、知情同意书中所提及的已知的或可预计的风险 已上市的药品说明书等信息 与所研究的疾病自然病程,受试者发生不良事件的转归等
2023年GCP培训考核试题及答案

2023年GCP培训考核试题及答案1 .本院发生的严重不良反应或严重不良事件需及时,即研究者获知后()小时通知本伦理委员会。
A.12B.24(正确答案)C.36D.482 .在疫情爆发等突发事件紧急情况下,一般在()小时内开展伦理审查,出具审查意见?A.72(正确答案)B.36C.24D.3个工作日3 .伦理审查委员会应当要求研究者提供审查所需材料,并在受理后()天内开展伦理审查并出具审查意见?A.10B.15C.60D∙30(正确答案)4 .本院新修订的《制定标准操作规程的标准操作规程》规定,机构文件系统颁布后()内,机构办负责组织各专业组研究成员对文件主要内容进行培训,保留培训记录。
A.10个工作日B.15个工作日C.20个工作日(D.1个月内5.本院新修订的《制定标准操作规程的标准操作规程》规定,()对文件进行常规的全面审核与修订更新。
A.每年B.每两年,C每三年D.每五年6.本院《药物临床试验安全性事件管理制度》规定,研究者在接收到申办者评估后的SUSAR报告,应及时签收阅读,并考虑是否对研究参与者的治疗采取相应的调整措施,并在接收到SUSAR报告后()递交至伦理办公室。
A.24小时内B.7个工作日C.14个工作日D.15天内7 .指研究参与者接受试验用药品后出现死亡、危及生命、永久或者严重的残疾或者功能丧失、研究参与者需要住院治疗或者延长住院时间,以及先天性异常或者出生缺陷等不良医学事件。
()A.严重不良事件(SAE)8 .可疑且非预期严重不良反应(SUSAR)C.不良事件D.不良反应9 .试验资料应保留多久?()A.2年B.3年C.4年D.应保存临床试验资料至临床试验终止后五年10 本院《临床试验人员培训制度》规定,机构办公室每年度至少组织一次医院内部培训()A.一次(B二次C.三次D.四次11 .试验用药品有哪些?()A.临床试验中的试验药B.对照药,工)C.安慰剂,D.申办方提供的伴随用药(,)IL根据药物与不良事件因果关系判断标准,将不良事件与受试药物应用的相关性包括()A.肯定有关(B.很可能有关I T)C.可能有关D.无法判断12.试验方案(Protocol),指说明临床试验()考虑和组织实施的文件。
研究者手册中安全性参考信息撰写技术指导原则

202112目录一、概述 (1)二、安全性参考信息的内容 (1)(一)预期严重不良反应 (1)(二)致死和/或危及生命的严重不良反应 (3)(三)因特异性和/或严重程度视为非预期的情形 (3)(四)安全性参考信息中不应包含的安全性信息 (5)三、安全性参考信息的呈现形式 (5)(一)位置 (6)(二)呈现形式 (6)(三)预期严重不良反应的术语 (8)(四)尚未发现预期严重不良反应的安全性参考信息 (9)四、安全性参考信息的适用版本 (9)五、安全性参考信息的变更 (9)六、安全性参考信息的质量管理体系 (10)七、安全性参考信息参考已上市药品说明书中不良反应的情形 (10)八、联合用药的安全性参考信息 (11)九、参考文献 (11)十、示例 (13)一、概述安全性参考信息(Reference Safety Information, RSI)通常是研究者手册(Investigator’s Brochure, IB)中的一个预期严重不良反应的列表。
申办者应根据RSI评估临床试验期间发生的所有可疑严重不良反应的预期性。
本指导原则旨在指导获准开展药物(包括中药、化学药及生物制品)临床试验的IB中RSI的撰写。
应用本指导原则时,请同时参考国际人用药品注册技术协调会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH)《E2A:临床安全性数据的管理:快速报告定义和标准》、《E2F:研发期间安全性更新报告》指导原则等。
本指导原则仅代表药品监管部门当前的观点和认识,不具有强制性的法律约束力。
随着科学研究的进展,本指导原则中的相关内容将不断完善与更新。
本指导原则为撰写安全性参考信息的一般考虑,尚不能涵盖所有情形。
如有未能阐明的个性化问题,可与药审中心进行沟通。
可疑且非预期严重不良反应审查

⑥此安全性事件是否应报告成SUSAR;
⑦采取了保护受试者人身安全和健康权益措施。
【否】:以上七要点的任意一点不合理时,应判断为【此安全性信息报告中的内容不合理】。
【不适用】:一般无此情况。
10、是否导致受试者继续参与试验的风险大于获益
风险程度建议考虑:
目前SUSAR的转归状态;
可疑且非预期严重不良反应审查
可疑且非预期严重不良反应审查报告表
项目类别
□药物□医疗器械□IIT□医疗新技术
项目名称
申办者/项目来源
研究专业
我院主要研究者
一、可疑且非预期的不良反应的情况
报告类型
□首次□随访□总结
SUSAR诊断
发生时间
报告时间
预期的判断
□预期□非预期
是否为SUSAR
口是口否
SUSAR程度
【否】;
导致医疗事故的情况。
【不适用】:一般无此情况。
8、安全性信息报告内容是否完整
【是】:法规要求SUSAR报告需包含七要素:
①患者详细资料,如姓名缩写等;
②可疑的药物,包括药物名称等;
③其他治疗,如合并用药等;
④可疑的药物不良反应的详细资料:如ADR严重程度等;
⑤事件报告人信息;
⑥申办者公司信息;
【否】指当前相关资料(如IB)所描述预期风险。
【否】指当前相关资料(如IB)所描述预期风险。
【不适用】:一般无此情况。
3、安全性事件是否与试验药物有关
【是】:指判断SUSAR与试验药物肯定有关、可能有关、无法评价。
【否】:指判断SUSAR与试验药物可能无关、肯定无关。
【不适用】:待评价情况(因资料)。
可疑医疗器械不良反应报告表范文

可疑医疗器械不良反应报告表范文英文回答:Adverse Event Reporting Form for Suspected Medical Device Adverse Reactions.Name: [Your Name]Date: [Date]Patient Information:Name: [Patient's Name]Age: [Patient's Age]Sex: [Patient's Gender]Medical History: [Brief summary of patient's medical history]Device Information:Device Name: [Name of the medical device]Manufacturer: [Name of the manufacturer]Model/Serial Number: [Model/Serial Number of the device]Date of Implantation/Use: [Date of deviceimplantation/use]Description of the Adverse Event:Please provide a detailed description of the adverse event experienced by the patient, including any signs or symptoms observed.Example: I recently used a blood pressure monitoring device on a patient. The patient experienced severe painand discomfort during the procedure. The device seemed tobe malfunctioning as it was displaying inconsistentreadings. The patient's blood pressure was also abnormally high, which was concerning.Actions Taken:Please describe any actions taken in response to the adverse event, such as discontinuation of device use, medical intervention, or device replacement.Example: As soon as I noticed the patient's discomfort and the device malfunction, I immediately stopped using the device. I informed the patient about the situation and provided them with alternative methods for monitoring their blood pressure. I also reported the incident to thehospital's medical device department for further investigation.Outcome:Please describe the outcome of the adverse event, including any medical interventions or treatments provided to the patient.Example: After discontinuing the use of the blood pressure monitoring device, the patient's pain and discomfort subsided. The patient was closely monitored for any further complications and was eventually switched to a different device for blood pressure monitoring. No further adverse events were reported.Preventive Measures:Please suggest any preventive measures that can be taken to avoid similar adverse events in the future.Example: To prevent similar adverse events in the future, it is important to conduct regular maintenance and calibration checks on medical devices. Additionally, healthcare professionals should receive proper training on device usage and troubleshooting. It is also crucial to establish a robust reporting system for adverse events to ensure prompt investigation and appropriate action.中文回答:可疑医疗器械不良反应报告表。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
附件
可疑且非预期严重不良反应(SUSAR)个案报告表首次报告□随访报告□编码:
申办者名称:地址:报告日期:年月日
注意事项:
一、此表供疫苗临床试验申办者向国家食品药品监督管理总
局药品审评中心报告SUSAR个案使用。
研究者报告严重不良事件的报告表由申办者确定,原则上至少应涵盖此报告表信息。
二、申办者以纸质和电子两种方式进行报告,其中纸质报告用于存档。
纸质报告须附临床试验批件复印件,加盖公章后快递,地址:北京市海淀区复兴路甲1号,收件人:国家食品药品监督管理总局药品审评中心资料组,邮编:100038。
电子报告可选择传真或电子邮箱,传真号码:,电子邮箱:。
资料组联系人:和渝红。
三、可疑不良反应指受试者在任何剂量下出现的与用药目的无关的有害反应,经分析认为与药物的关系是至少可能相关。
四、非预期不良反应指在性质、程度、后果或频率方面,与先前方案或其他相关资料(如研究者手册等文件)所描述的预期风险不一致的不良反应。
研究者手册是用于判断某不良反应是否预期或非预期的主要参考文件。
如:(1)急性肾衰在研究者手册中列为可能的不良反应,但试验过程中出现间质性肾炎,即应判断为非预期不良反应;(2)肝炎在研究者手册中列为可能的不良反应,但试验过程中发生爆发性肝炎,即应判断为非预期不良反应。
五、严重不良事件/反应指以下情形之一的事件/反应:(1)导致死亡;(2)危及生命;(3)致癌、致畸、致出生缺陷;(4)导致伤残或器官功能损伤、影响工作能力;(5)导致住院或延长住院时间;(6)导致其他重要医学事件,如不进行治疗可能出现上述所列情况的。
六、其他明显影响试验药物风险效益评估的信息或可能影响
用法用量或影响总体研发策略的信息,按“潜在严重安全性风险的信息”处理,进行个案报告。
如:预期严重不良反应发生率增加且具有临床意义,对受试者有明显危害,试验疫苗被判断无效,最近完成的非临床研究发现重大安全性问题(如致癌性等)等。
七、研究者或申办者首次获知当天为第0天。
临床试验结束或随访结束后至获得审评审批结论前发生的严重不良事件,研究者需及时报告申办者,并由申办者判断是否符合本规定的个案报告条件,符合条件的应报告。
八、盲法试验中发生严重不良事件时,为便于判断严重不良事件与试验疫苗的相关性,申办者和研究者可协商是否对个别受试者揭盲。
如研究者揭盲,即认为申办者获知该受试者使用疫苗情况。
当严重不良事件为主要疗效终点时,不建议申办者以个案形式向食品药品监督管理部门报告。
九、申办者应及时将阳性对照疫苗相关的严重不良反应报告给该疫苗生产商,由该疫苗生产商根据相关规定报告上市后疫苗不良反应监测机构。