粗粒化分子动力学-力场开发与应用
免疫学检测中抗体固定化方法的研究现状

免疫学检测中抗体固定化方法的研究现状刘爱平;申文浩;王小红;李诚【摘要】Immunological assays are widely applied in food safety and environmental monitoring because they are rapid, sensitive and of low price. Antibody, the core reagent in immunological assays, usually needs to be immobilized to capture antigen. It is of great significance to improve detection sensitivity through effective immobilization of antibody. This review seeks to overview the characteristics and application of each kind of immobilization methods in immunological assays, and the developing trends of oriented-immobilization technique were prospected.%免疫学检测技术,因具有快速、灵敏、成本低等优点,被广泛应用于食品安全、环境监测等领域.抗体是免疫学检测的核心试剂,免疫学检测中常需要对抗体进行固定,高效固定抗体对提高检测灵敏度具有重要意义.本文对抗体固定化各类方法的特点及其在免疫学检测中的应用进行综述,并对抗体定向固定化技术发展趋势进行了展望.【期刊名称】《江苏农业学报》【年(卷),期】2017(033)003【总页数】7页(P714-720)【关键词】抗体;定向固定;生物素-亲和素;亲和肽配基;DNA引导固定法【作者】刘爱平;申文浩;王小红;李诚【作者单位】四川农业大学食品学院,四川雅安 625014;四川农业大学食品学院,四川雅安 625014;华中农业大学食品学院,湖北武汉 430070;四川农业大学食品学院,四川雅安 625014【正文语种】中文【中图分类】Q599免疫学检测技术,因具有快速、灵敏、成本低、适合大批量检测等优点,被广泛应用于食品安全、环境监测、疾病诊断等领域[1-3]。
一种基于Martini力场的色素炭黑粗粒化模型建立方法[发明专利]
![一种基于Martini力场的色素炭黑粗粒化模型建立方法[发明专利]](https://img.taocdn.com/s3/m/7527bb5ca88271fe910ef12d2af90242a895ab30.png)
(19)中华人民共和国国家知识产权局(12)发明专利申请(10)申请公布号 (43)申请公布日 (21)申请号 202110128421.2(22)申请日 2021.01.29(71)申请人 太原理工大学地址 030024 山西省太原市迎泽西大街79号(72)发明人 徐娜 刘子璐 吕耀东 张瑾渊 贺文云 (74)专利代理机构 太原高欣科创专利代理事务所(普通合伙) 14109代理人 冷锦超 邓东东(51)Int.Cl.G16C 10/00(2019.01)G06F 30/25(2020.01)G06F 119/14(2020.01)(54)发明名称一种基于Martini力场的色素炭黑粗粒化模型建立方法(57)摘要本发明提出一种基于Martini力场的色素炭黑粗粒化模型建立方法,属于纳米化学计算技术领域;具体是将球形色素炭黑粗粒化模型分为了中心珠子以及最外层球壳形珠子,在Martini力场中定义两类型珠子,将摩尔质量、势作用参数进行赋值,在中心珠子以及最外层球壳形珠子的连接上采用键连接,以保证进行分子动力学模拟时可以不变小,不坍缩;由于整个粗粒化模型只有最外层球壳形珠子以及中心珠子,这大大减少了珠子的数量,可以在保证分子动力学计算的准确性同时大大的降低计算量,提高了计算的速度,也避免了计算上的浪费。
权利要求书1页 说明书3页 附图2页CN 112768007 A 2021.05.07C N 112768007A1.一种基于Martini力场的色素炭黑粗粒化模型建立方法,其特征在于,包括以下步骤:S1、根据所需色素炭黑粒径大小,结合色素炭黑平均密度,计算所需色素炭黑的质量;S2、将色素炭黑分为中心珠子和以中心珠子为球心,均匀排布在球面上的最外层珠子;中心珠子和最外层珠子之间、最外层珠子之间均为键连接;根据色素炭黑粒径大小,确定最外层珠子数量,将所需色素炭黑的质量平均分配给最外层珠子;S3、根据色素炭黑粒径大小,确定中心珠子与最外层珠子之间的键长、最外层珠子之间的键长,以及为了维持结构刚性所需要的键能参数;S4、在Martini力场中分别定义中心珠子以及最外层珠子类型,根据中心珠子和最外层珠子的摩尔质量大小修改所定义珠子在Martini力场中的各类参数。
MaterialsStudio软件

MaterialsStudio软件一、Materials Studio软件的主要应用领域包括:金属材料研究无机非金属材料研纳米材料研究高分子及其复合材料研究表界面研究化学反应研究含能材料研究生物、医药研究在晶体结构、形貌研究中的应用QSAR 的应用Perl 语言的应用Accelrys(美国)公司是世界领先的计算科学公司,是一系列用于科学数据的挖掘、整合、分析、模建与模拟、管理和提交交互式报告的智能软件的开发者,是目前全球范围内唯一能够提供分子模拟、材料设计、化学信息学和生物信息学全面解决方案和相关服务的软件供应商,所提供的全面解决方案和科技服务满足了当今全球领先的研究和开发机构的要求。
Materials Studio多尺度分子模拟平台是Accelrys公司(美国)在材料设计领域的核心产品。
它融合多种模拟方法,整合多达23 个功能模块,实现从电子结构解析到宏观性能预测的全尺度科学研究。
在国内拥有近400家用户,分布在石油、化工、环境、能源、制药、电子、食品、航空航天和汽车等工业领域和教育科研部门;相关的研究工作在Nature、Science等各类权威期刊上发表论文过万篇。
Materials Studio分子模拟软件采用了先进的模拟计算思想和方法,如量子力学(QM)、线性标度量子力学(Linear Scaling QM)、分子力学(MM)、分子动力学(MD)、蒙特卡洛(MC)、介观动力学(MesoDyn)和耗散粒子动力学(DPD)、统计方法QSAR(Quantitative Structure - Activity Relationship )等多种先进算法和X射线衍射分析等仪器分析方法;同时产品提供了界面友好的的模拟环境,研究者能方便地建立三维结构模型,并对各种小分子、纳米团簇、晶体、非晶体以及高分子材料的性质及相关过程进行深入的研究,得到切实可靠的数据。
Materials Studio分子模拟软件支持Windows和Linux操作平台,用户可以自由定制、购买自己的模拟方法和模块,以满足特定领域研究需求。
分子动力学在沥青中的应用

分子动力学在沥青中的应用摘要:对于沥青再生技术而言,再生中新旧沥青的混溶过程是一个关键问题,其直接影响着合适的新沥青胶结料等级及其用量的确定,并对保障沥青再生效果及沥青路面的路用性能具有重要意义。
为此本文采用分子动力学方法,从分子尺度构建新旧沥青混溶模型,在微观模拟新旧沥青混溶过程的同时,计算分析再生沥青的宏观物理性能。
关键词:再生沥青;分子动力学;新旧沥青混溶;再生剂1.绪论1.1.研究背景和意义公路作为一项为公众提供出行便利性的基础设施,一直是国家建设的重点工程,是衡量一个国家经济水平和发展状况的标志之一。
目前我国公路的主要路面类型为沥青混凝土路面,约占我国公路路面总面积的80%,而随着我国交通运输业的不断发展及公路的不断使用,已经有很大一部分的沥青路面面临着养护维修乃至重建,甚至有些路面在短期内就已经受到了定程度的损坏,需要进行养护以延长使用寿命。
这就导致了每年将产生大量的废旧沥青混合料。
在沥青混合料再生技术中,一个关键问题是再生过程中新旧沥青混合问题,其混合程度直接影响再生混合料的性能指标,对整个沥青混合料的性能指标有直接的影响,旧沥青是否完全参与再生,对于新沥青的等级、掺量、混合料设计等一系列问题起基础性的作用。
研究新旧沥青混合问题对沥青混合料再生技术有着重大的意义,是再生理论的基础性内容之一,对再生体系的完善起促进作用。
目前关于新旧沥青混溶状态的研究大多集中在宏观实验研究,但由于沥青的分子组成十分繁复,新旧沥青混溶的过程也极为复杂,宏观实验难以较好地表征新旧沥青的混溶状态,一些微观试验手段例如原子力显微镜、扫描电镜等,虽然可以观察到微观始末的状态,但其内部结构的变化过程却难以观测。
因此,分子动力学是研究新旧沥青混溶状态一个直接有效的研究手段。
而分子动力学通过定义粒子支架相互作用,通过对体系内分子运动进行计算,来得到体系的热力学性质。
利用分子动力学研究新旧沥青混溶状态,在对分子微观行为进行研究的同时,也可以计算物质的宏观物理性质。
第四章 分子动力学

分子动力学与分子力学不同,它求解的是随时间变化的分子的状态、行为和过程。
分子动力学将原子看作为一连串的弹性球,原子在某一时刻由于运动而发生坐标变化。
在运动的任一瞬间,通过计算每个原子上的作用力和加速度,来测定它们的位置和运动速度。
由于一个原子的位置相对于其他原子的位置不断变化着,同时力也在变化,可用适当的力场方法,通过评价体系的能量,计算出任一特定原子的力。
分子动力学模拟可作瞬时的、通常为皮秒级(10-12s)的分析,由此模拟计算而获得以一定位置和速度存在的原子的运动轨迹。
计算中根据分子体系的大小、特点和要求来决定模拟时间的长短。
分子动力学方法是一通用的全局优化低能构象的方法。
用分子动力学模拟可使分子构象跨越较大的能垒,因此可以通过升温搜寻构象空间,势能的波动对应着分子构象的变化,当总能量出现最小值时,在常温下(300K)平衡,即可求得低能构象。
在常温下的分子动力学模拟需要很长的时间来克服能量势垒,因此分子动力学对分子构象空间的取样相当缓慢。
提高分子体系的温度,可加大样本分子构型空间的取样效率。
分子动力学计算中,常使用蒙特卡洛算法和模拟退火算法。
蒙特卡洛算法:是一种统计抽样方法。
其基本思想是在求解的空间中随机采样并计算目标函数,以在足够多的采样点中找到一个较高质量的最优解作为最终解。
在动力学计算全局优化低能构象时,以经验势函数随机抽样,不断抽取体系构象,使其逐渐趋于热力学平衡。
该方法需要大量采样才能得到较精确的结果,因此收敛速度较慢。
模拟退火算法:退火是将金属或其他固体材料加热至熔化后,再非常缓慢地冷却的过程。
缓慢冷却是为了凝固成规则的处于最稳态的坚硬晶体状态。
模拟退火算法用于分子动力学计算时,可有效地求得分子的全局优势构象。
过程为:先使体系升温,在高温下进行分子动力学模拟,使分子体系有足够的能量,克服柔性分子中存在的各种旋转能垒和顺反异构能垒,搜寻全部构象空间,在构象空间中选出一些能量相对极小的构象;然后逐渐降温,再进行分子动力学模拟,此时较高的能垒已无法越过,在极小化后去除能量较高的构象,最后可以得到相应的能量最小的优势构象。
【国家自然科学基金】_粗粒化模型_基金支持热词逐年推荐_【万方软件创新助手】_20140731
推荐指数 2 2 1 1 1 1 1 1 1 1 1 1 1 1 1
2012年 序号 1 2 3 4 5 6 7 8 9 10 11 12
科研热词 分子动力学模拟 表面活性剂 结晶行为 粗粒化模型 粗粒化投影积分 粗粒化 电化学性能 多尺度模拟 固体氧化物燃料电池 半晶态聚合物 力场 martini
推荐指数 2 1 1 1 1 1 1 1 1 1 1 1
2013年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
2013年 科研热词 耗散粒子动力学 设计思维 计算机辅助草图设计 表面活性剂 草图设计 草图行为 自组装 聚甲基丙烯酸甲酯 纳米复合物 纳注射成型 红细胞 粗粒化模型 粗粒化力场 球形胶束 渗流转变 数值模拟 微乳胶粒 变形 双亲无规共聚物 创意拐点 分形维数 分子模拟 分子动力学模拟 分子动力学 亲水性 推荐指数 2 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
2014年 序号 1 2 3 4 5 6 7 8 9 10 11 12 13
科研热词 非晶态聚合物 耗散粒子动力学模拟 结构塌缩 粗粒化模型 相行为 核酸单链 拥挤大分子 变形机理 半晶态聚合物 分子动力学模拟 偶分布函数 t形三组分双亲分子 monte carlo模拟
推荐指数 1 1 1 1 1 1 1 1 1 1 1 1 1
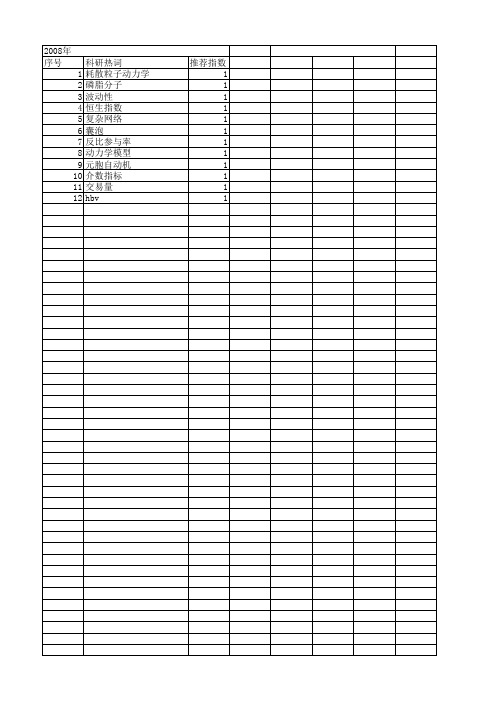
2008年 序号 1 2 3 4 5 6 7 8 9 10 11 12
科研热词 耗散粒子动力学 磷脂分子 波动性 恒生指数 复杂网络 囊泡 反比参与率 动力学模型 元胞自动机 介数指标 交易量 hbv
推荐指数 1 1 1 1 1 1 1 1 1 1 1 1
【CN110097927A】基于分子动力学测试电场作用下离子扩散系数的方法【专利】

(19)中华人民共和国国家知识产权局(12)发明专利申请(10)申请公布号 (43)申请公布日 (21)申请号 201910387260.1(22)申请日 2019.05.10(71)申请人 青岛理工大学地址 266033 山东省青岛市市北区抚顺路11号(72)发明人 侯东帅 张庆恩 李萌萌 王攀 王鑫鹏 (74)专利代理机构 北京志霖恒远知识产权代理事务所(普通合伙) 11435代理人 朱昀(51)Int.Cl.G16C 10/00(2019.01)G16C 20/30(2019.01)(54)发明名称基于分子动力学测试电场作用下离子扩散系数的方法(57)摘要本发明属于微观纳米材料技术领域,尤其涉及基于分子动力学测试电场作用下离子扩散系数的方法,包括以下步骤:基于分子动力学仿真软件Materials Studio构建水化硅酸钙(C -S -H)模型,导出car,cor文件;基于大规模原子分子并行模拟器(lammps)导出data文件;LJ势函数用于估计分子的大小及非键结作用势能的最低值,求出不同原子之间的LJ势作用函数;撰写in文件,运行in文件;给模型在X方向施加电场强度大小分别为计算不同电场大小下的均方位移值;输出cl.msd文件;根据爱因斯坦的扩散定律通过Matlab分析以及Origin做图,得到离子的扩散系数。
本发明能快速、准确、无污染地预测混凝土的氯离子扩散系数。
权利要求书2页 说明书5页 附图3页CN 110097927 A 2019.08.06C N 110097927A1.基于分子动力学测试电场作用下离子扩散系数的方法,其特征在于,包括以下步骤:步骤1、用分子动力学仿真软件Materials Studio构建C-S-H模型,也就是水化硅酸钙模型;分别给钠离子、氯离子、钙离子、硅氧四面体中的硅原子以及氧原子赋电荷,保持电荷平衡,电荷平衡时为0,采用CVFF力场能量最小化保证模型模拟时能量最低,Materials Studio最后导出car,cor文件,把导出的car,cor文件复制到lammps中;ClayFF力场中使用SPC模型来表示水分子,利用Harmonic势场函数来分别表示O-H共价键的拉伸、H-O-H角度的弯曲作用:E bond stretch ij=K1(r ij-r0)2,E angle bend ijk=K2(θijk-θ0)2,K1,K2分别表示拉伸和扭转刚度,r0代表O-H键的平衡长度,θ0表示H-O-H的平衡角度,r ij 是为O和H原子间的分离距离,θijk是金属氧和氢的键角;步骤2、基于大规模原子分子并行模拟器lammps导出data文件,期间采用ClayFF力场中规定的原子参数设置模型各原子键角,键长强度值并根据ClayFF力场中明确的参数分别赋给data文件中钠离子、氯离子、钙离子、硅氧四面体中的硅原子和氧原子不同的势能值ε,以及钠离子、氯离子、钙离子、硅氧四面体中的硅原子以及氧原子之间的平衡距离δ;LJ势函数用于估计模型中所有原子的大小及非键结作用势能的最低值,ClayFF力场使用LJ势函数来表示钠离子、氯离子、钙离子、硅氧四面体中的硅原子和氧原子间的范德瓦尔斯力,模拟真实状态的模型中所有原子间作用,表达式为:其中r ij为i,j两个原子间距离,i,j为涉及模拟体系中发生阴阳离子之间相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子,D0,ij、R0,ij是C-S-H模型与观测到的结构和物理特性数据拟合得到的经验参数,分别表示i、j两个原子之间的能量与距离常数,i,j为涉及模拟体系中发生阴阳离子之间相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子;模拟过程中,离子之间存在库伦作用力,其静电库伦作用表达式为:式中,q i,q j表示模拟体系中相互作用的两种原子i,j各自的部分电荷;e是电子的电荷;r ij为i,j原子之间的分离距离;i,j为涉及模拟体系中发生阴阳离子之间相互作用的钠离子、氯离子、钙离子或硅氧四面体中的氧原子;ε0为真空的介电常数,值为8.8542*10-12F/m;计算值ε和δ查阅参考文献Clayff力场获得;步骤3、撰写in文件,运行in文件包括四部分:(1)C-S-H模型沿三维坐标X轴、Y轴、Z轴施加周期性边界;(2)固定C-S-H模型最底层原子,让溶液离子跑1000ps达到均匀分布;(3)解除C-S-H基底,让溶液和基底充分驰豫,分别给C-S-H模型中X轴,Y轴,Z轴方向分别赋初始力值大小为0,0,0;(4)设定lammps模拟参数:在in文件中,采用NVT系综,时间步长1fs,温度设定300K让整个盒子驰豫达到平衡,保证氯化钠溶液在C-S-H模型中扩散均匀,设定电场大小,以及电场方向;步骤4、给C-S-H模型在x轴方向或者y轴方向施加电场,继续跑2000ps,让离子在电场作用下运动,最后输出2000ps的原子轨迹进行matlab编程分析,即可计算不同电场大小下的均方位移大小,即MSD的大小;步骤5、lammps运行结束,输出均方位移文件cl.msd文件,均方位移文件为原子在电场作用下充分驰豫,平衡后输出的氯离子模拟数据;步骤6、根据爱因斯坦的扩散定律通过Matlab分析以及Origin做图,得到离子的扩散系数D(t)。
分子动力学模拟的若干基础应用和理论

分子动力学模拟的若干基础应用和理论一、本文概述分子动力学模拟是一种基于经典力学的计算方法,通过求解分子体系的牛顿运动方程,模拟分子在特定条件下的动态行为。
该方法广泛应用于物理、化学、生物和材料科学等领域,为研究者提供了一种有效的工具,以深入理解和预测分子系统的宏观性质。
本文旨在探讨分子动力学模拟的若干基础应用和理论,从基础概念出发,阐述其基本原理、模拟方法以及在各个领域中的应用实例。
我们将详细介绍分子动力学模拟的核心技术,包括力场模型、初始条件设定、积分算法和模拟结果的解析等。
本文还将讨论分子动力学模拟的局限性以及未来的发展方向,以期为相关领域的研究人员提供有益的参考和启示。
二、分子动力学模拟的理论基础分子动力学模拟(Molecular Dynamics Simulation, MDS)是一种强大的计算技术,通过求解分子体系的牛顿运动方程,模拟分子在特定条件下的动态行为。
其理论基础主要建立在经典力学、统计力学以及量子力学之上,但在大多数应用中,由于计算能力的限制,经典力学是主要的工具。
在经典力学中,每个分子的运动可以通过牛顿第二定律来描述,即力等于质量乘以加速度(F=ma)。
在分子动力学中,这些力通常是分子间相互作用力,包括范德华力、氢键、库仑力等。
这些力可以通过分子力学模型或量子力学方法计算得出。
分子动力学模拟通常包括以下几个主要步骤:需要设定模拟的初始条件,包括分子的初始位置、速度和模拟的温度、压力等环境参数。
然后,根据分子间的相互作用力,通过求解牛顿运动方程,计算出每个分子在下一时刻的位置和速度。
这个过程会不断重复,直到模拟达到预设的时间长度或达到某种平衡状态。
在模拟过程中,为了处理大量的分子和长时间的模拟,通常会采用一些近似和简化的方法,如截断半径、周期性边界条件等。
由于分子间的相互作用力往往非常复杂,因此在模拟中通常会采用一些经验性的力场模型,如Lennard-Jones势、Morse势等。