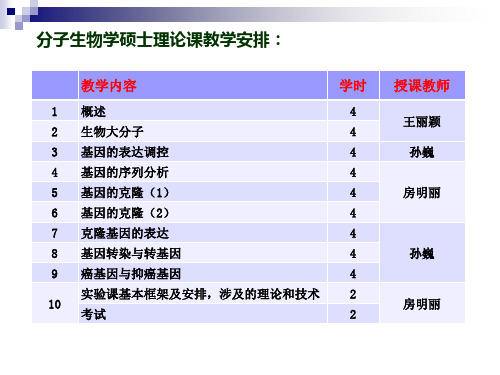
Gene 序列分析
基因的序列分析 20131024
双脱氧核苷酸(ddNTP)
3’-5’磷酸 二酯键
不能与下一个脱氧 核苷酸结合!
(1)Sanger双脱氧末端终止法
背景知识
在PCR反应体系中,如果只加入一条引物是什么样子的结果?
• •
单引物只能扩增单链DNA 扩增的包含引物的单链DNA 不对称PCR (asymmetric PCR) 是用不等量的一对引物,PCR 扩增后产生大量的单链 DNA(SSDNA).
KRAS基因突变主要发生在密码子12,13上
密码子12/13发生变异的患者
应用 (举例说明: 应用焦磷酸测序法检测DNA甲基化)
焦磷酸测序法检测DNA甲基化
5’甲基胞嘧啶 在亚硫酸盐的作用下变成胸腺嘧啶
焦磷酸测序可在一次检测中快速定量一个或多个甲基化位点 焦磷酸测序技术可检测宫颈癌中UTF1启动子区域甲基化水平
将电泳分离的待测基因组DNA酶切片段转移到一定的固
相支持物上,然后与标记的核酸探针进行杂交的过程, 基本流程如下:
①制备待测 DNA 样品、标记基因探针;
②电泳分离待测DNA样品; ③待测DNA样品的变性、转膜;
④杂交;
⑤显色。
Southern 印迹杂交
Southern印迹基本操作过程
两种特殊底物 APS, 荧光素 四种酶: • DNA聚合酶 • ATP硫酸化酶• 荧光素酶
• 三磷酸腺苷双磷酸酶
(3)焦磷酸测序法
原理
DNA聚合酶
APS+
硫酸化酶 荧光素酶
双磷酸酶
荧光素+
(3)焦磷酸测序法
测序原理
第一步:加入测序引物,相关酶,底物,和其他试剂 第二步:每次加入一种dNTP,如果结合,则会产生一个焦磷 酸(PPi) 第三步:硫酸化酶转化PPI为ATP, ATP使荧光素酶发出荧 光。(产生的荧光强度与结合的核苷酸成正比) 第四步:多余的dNTP被降解,开始新一个循环。 看一下视频
生物信息学中的基因序列分析与预测

生物信息学中的基因序列分析与预测生物信息学是一门综合学科,它将计算机科学、数学和统计学等技术应用于生物学领域。
基因序列分析与预测是生物信息学中的重要研究领域之一,它涉及到对基因序列的分析、注释和预测。
基因序列是生物体内以DNA或RNA形式存在的遗传信息。
通过对基因序列的分析,我们可以了解基因的功能和结构,进而深入研究生物体的生理过程和疾病发生机理。
基因序列的注释则是对基因序列进行功能和结构的解读和标记,在基因组学研究和生物学研究中起到关键作用。
基因序列的预测是通过生物信息学技术对未知基因序列进行功能和结构的预测。
在基因组学研究中,大量基因序列还没有被准确注释,因此基因序列的预测对于深入研究生物体的特征和功能非常重要。
基因序列预测可以通过多种算法和技术来实现,其中最常用的方法包括序列比对、开放阅读框(ORF)预测、蛋白质结构预测等。
序列比对是基因序列分析的基本方法之一,它通过比较待分析序列与已知序列数据库中的序列进行比较,从而找到相似的区域和序列特征。
根据比对结果,可以判断待分析序列与已知序列的亲缘关系、功能和结构等信息。
开放阅读框(ORF)预测是对基因序列中的蛋白编码区域进行预测。
开放阅读框是指在核苷酸序列中没有起始密码子和终止密码子的连续核苷酸序列。
通过使用启动子预测算法和终止密码子识别算法,可以准确地预测基因序列中的开放阅读框,进而推断蛋白编码区域的位置和功能。
蛋白质结构预测是预测待分析基因序列所编码的蛋白质的三维结构。
蛋白质的结构对于其功能和相互作用非常关键,因此准确地预测蛋白质结构对于研究蛋白质的功能和疾病发生机制具有重要意义。
蛋白质结构预测方法主要分为比较模型和折叠模型两种,通过比对已知结构的同源蛋白质,或者通过物理化学规则和算法,可以预测待分析蛋白质的结构。
在生物信息学中,基因序列分析与预测常常是多领域合作的结果,涉及到计算机科学、生物学、数学和统计学等多学科的知识与技术的融合。
随着高通量测序技术的不断发展,我们可以获取到大量的基因序列数据,这为基因序列分析与预测提供了更多的机会和挑战。
生物信息学中基因序列分析方法及注意事项

生物信息学中基因序列分析方法及注意事项在生物信息学领域中,基因序列分析是一项重要的研究任务。
基因序列分析可以帮助我们理解基因组的结构和功能,寻找潜在的基因组变异,并预测基因的功能。
本文将介绍一些常见的基因序列分析方法,并提供一些建议和注意事项。
一、基因序列比对方法基因序列比对是将一个基因序列与一个或多个参考序列进行比较的过程。
比对的目的是识别序列中的保守元素以及识别已知序列与未知序列之间的相似之处。
常见的基因序列比对方法包括全局比对和局部比对。
1.全局比对:全局比对方法适用于两个序列之间具有较高的相似性。
其中最常用的方法是Smith-Waterman算法,该算法可以找到两个基因序列之间的最佳比对结果,包括匹配、替代和间隔。
2.局部比对:局部比对方法适用于寻找两个序列之间的片段相似性。
著名的局部比对算法有BLAST和FASTA。
这些方法能够快速识别目标序列中与参考序列相似的片段,并生成比对结果。
二、基因预测方法基因预测是指根据DNA序列推断基因的位置和结构。
基因预测的主要挑战在于标识和区分编码蛋白质的基因和非编码区域。
以下是一些常用的基因预测方法:1.基于序列特征的预测:该方法使用DNA序列中的特定序列特征来识别编码蛋白质的基因。
这些特征包括启动子序列、剪切位点、启动密码子和终止密码子等。
通过在目标序列中搜索这些特征,可以预测基因的位置和结构。
2.基于比对的预测:该方法将已知的蛋白质序列与目标序列进行比对,从而识别可能的编码蛋白质的区域。
该方法依赖于已知蛋白质序列的数据库,如GenBank和SwissProt。
三、基因表达分析方法基因表达分析是通过测量RNA或蛋白质的产量来研究基因在不同组织、生长阶段或环境条件下的表达。
以下是一些常见的基因表达分析方法:1.转录组测序(RNA-seq):该方法使用高通量测序技术直接测量基因转录产物(mRNA)的产量。
通过RNA-seq技术,可以发现新基因、检测剪接异构体和检测SNP等。
遗传学研究中基因序列的分析方法

遗传学研究中基因序列的分析方法随着DNA测序技术的不断发展以及基因组学领域的兴起,基因序列分析已经成为生命科学研究中的重要一环。
基因序列分析为研究基因的功能和结构奠定了基础,也为了解基因在遗传疾病中的作用提供了必要的手段。
本文将介绍常见的基因序列分析方法和工具,以及它们在遗传学研究中的应用。
1. 基因注释基因注释是将分子生物学实验数据和计算机分析结果结合起来,确定基因区的位置,预测基因的结构和功能。
基因注释包括以下几个方面:1.1 基因定位基因定位是基于物理或遗传地图来确定某个基因位于哪条染色体上的一种方法。
目前最常用的基因定位方法是PCR-RFLP(PCR-Restriction Fragment Length Polymorphism)和AFLP (Amplified Fragment Length Polymorphism)。
PCR-RFLP是将多态性位点PCR扩增,再用特异的内切酶切割,根据DNA片段长度的不同,确定是否存在基因多态性。
AFLP则是采用粘性末端技术,先用不同长度的引物对DNA片段进行PCR扩增,然后将扩增产物限制性切割,再用凝胶电泳进行分离。
基因定位是基因注释的重要步骤,由此可以推算出大部分疾病的基因所在位置。
1.2 基因结构预测基因结构预测是根据基因组序列预测基因结构的一种方法。
目前,最常用的基因结构预测程序有GlimmerHMM、GeneMark、FGENESH、GENSCAN和AUGUSTUS等。
它们都是基于概率模型的软件,通过分析DNA序列的k-mer频率、AT含量等特征,确定起始密码子、终止密码子位置和剪接位点等关键信息。
1.3 基因功能预测基因功能预测是通过转录组和蛋白质组数据,预测基因在生物过程中的功能以及参与的通路。
这里我们介绍两种基因功能预测方法:GO分析和KEGG通路分析。
GO(Gene Ontology)分析是一种基于语义化的功能分析方法,可以揭示基因及其编码产物的功能、过程和位置。
生物信息学中的基因序列分析方法研究

生物信息学中的基因序列分析方法研究【引子】随着基因测序技术的飞速发展,我们进入了一个基因数据爆炸的时代。
在海量的基因组数据中,如何从中提取有关生物信息?生物信息学的发展成为解决这一问题的重要工具。
基因序列分析是生物信息学中的核心内容之一,本文将从多个角度探讨生物信息学中的基因序列分析方法的研究。
【基因序列分析方法的介绍】基因序列分析方法是一种运用计算机和数学算法对基因序列进行统计、分析、解读和预测的方法。
这些方法旨在揭示基因与生命现象之间的关联,为生命科学研究提供依据。
基因序列分析方法广泛应用于基因功能预测、基因家族鉴定、物种鉴定、基因演化和疾病研究等领域。
【基因序列比对方法】基因序列比对是分析基因序列中的相似性和差异性的基础方法。
以蛋白质序列比对为例,最常用的算法是BLAST(基本局部比对搜索工具)。
BLAST通过比对查询序列与数据库中的序列,找出两者之间的相似性。
BLAST算法的独特之处在于采用了多种启发式搜索策略,极大地提高了比对速度。
而对于DNA序列比对,常用方法有Smith-Waterman算法和Needleman-Wunsch算法。
这些方法都对序列进行全比对或半全比对,是基因序列分析的重要基石。
【基因结构预测方法】基因结构预测是确定基因组中基因的位置、外显子和内含子的边界以及剪接位点的方法。
它在解析基因功能、开展基因组注释和研究剪接变异等方面扮演着重要角色。
目前,常用的基因结构预测方法主要分为三大类:比对依赖方法、剪接位点预测方法和贝叶斯统计方法。
比对依赖方法通过比对已知基因和测定位点的数据,推测新的位点和未知基因的位置。
剪接位点预测方法基于序列信息探测剪接异构体,代表性的算法包括MaxEntScan和SplicePort。
贝叶斯统计方法则通过概率模型研究剪接位点的概率,如BPP和GeneID。
这些方法的使用范围和精度各不相同,研究人员需要根据具体问题灵活选择。
【基因组注释方法】基因组注释是基因组中各个元件的功能和结构的描述,涉及外显子、内含子、启动子、转录因子结合位点等多个方面。
生物信息学中的基因序列分析技术解析

生物信息学中的基因序列分析技术解析生物信息学是一门综合学科,将生物学、计算机科学和统计学等领域的知识相结合,致力于从大规模的生物学数据中提取有用的信息和知识。
基因序列分析是生物信息学中的重要研究内容之一,通过对基因组中的DNA序列进行分析,可以揭示基因的结构、功能和调控机制。
本文将对生物信息学中的基因序列分析技术进行深入解析。
一、基因序列获取在进行基因序列分析之前,首先需要获得待分析的基因序列。
目前,基因序列获取的主要方法是基于高通量测序技术的方法,如Sanger测序、二代测序和三代测序。
1. Sanger测序Sanger测序是一种经典的测序方法,基于链终止法原理。
该方法通过引入低浓度的二进制链终止剂,使DNA合成过程中的链终止在不同的碱基位置。
然后,使用聚丙烯酰胺凝胶电泳将不同长度的DNA片段分离出来,并根据电泳结果确定序列。
尽管Sanger测序方法准确可靠,但速度较慢,无法满足高通量测序的需求。
2. 二代测序二代测序技术是目前广泛应用的高通量测序技术,包括 Illumina的测序技术、Ion Torrent的测序技术等。
这些技术采用了片段拼接和PCR扩增的方法,将DNA样本分割成小片段,并使用高度并行的测序反应同步测序。
这种高通量测序技术具有快速、成本低廉和数据量大等优点,为后续的基因序列分析提供了强大的数据支持。
3. 三代测序三代测序技术相比于二代测序技术具有更高的读长,能够直接测序较长的DNA分子。
代表性的三代测序技术有Pacific Biosciences (PacBio)和Oxford Nanopore Technologies(ONT)的测序技术。
这些技术主要基于单分子测序原理,通过测量单个DNA分子的链延伸或通过测量基于纳米孔的离子电流来进行测序。
三代测序技术的发展为更好地解析复杂的基因组结构和重复序列提供了可能。
二、基因序列比对基因序列比对是生物信息学中的重要任务,它主要通过将待分析的基因序列与已知参考序列进行比较,从而确定相似性和差异性。
基因序列分析与注释的研究方法

基因序列分析与注释的研究方法基因序列分析和注释是现代生物学领域中的重要研究方法。
随着科技的不断进步和创新,生物学研究方法也在不断地发展和完善。
在这些方法中,基因序列分析和注释是非常重要的,它们可以帮助我们更好地理解和预测生物的遗传特征。
一、基因序列分析基因序列分析是指首先获取DNA序列,然后对该序列进行分析。
基因序列分析通常涉及到以下的几个方面:基因组比较和分析、拼接和修补、序列质量控制,基因组注释等等。
1. 基因组比较和分析基因组比较和分析是指将两个或多个基因组的序列进行比较,以研究它们之间的相同性或差异性。
比较的方法包括比较DNA的一般结构和功能序列的相似性。
基于比较分析,可以得出生物分类、进化和种群分布等方面的结论。
2. 拼接和修补拼接和修补是指将多个不完整的DNA序列拼接成一条完整的序列,以便进一步的分析。
这个过程需要通过软件和算法来完成,其中最常用的是基于De Bruijn 图的算法。
3. 序列质量控制序列质量控制是指对不同的序列进行质量检测和控制,以确保数据的准确性和可靠性。
序列质量控制的方法包括使用质量峰(Phred)分值、去除低质量序列和过滤跨越边界的序列等。
二、基因组注释基因组注释是指对基因组序列进行注释,以确定基因的结构、功能和表达。
基因组注释通常包括以下内容:基因预测、基因定位、可变剪接分析、调控元件注释等。
1. 基因预测基因预测是指通过软件和算法对未知的DNA序列进行分析,以确定哪些区域是编码基因。
基因预测提供了对基因组序列功能的了解,同时也是细胞和组织分化以及人类疾病研究的重要基础。
2. 基因定位基因定位是指将基因的位置比对到已知的染色体上,以确定基因在基因组中的位置。
基因定位是研究基因功能和疾病遗传学的基础。
3. 可变剪接分析可变剪接是指同一个基因在不同的组织和环境中通过不同的剪接方式产生不同的mRNA,并通过翻译产生不同的蛋白质。
可变剪接分析可以帮助我们更好地了解基因组的功能,并研究疾病在不同组织中的表达。
基因序列的分析与生物信息学研究

基因序列的分析与生物信息学研究近年来,基因序列分析与生物信息学研究在生物学领域中起到了非常重要的作用。
无论是从基础科研到应用研究,都离不开这些技术的支持。
本文将重点介绍基因序列分析与生物信息学研究的基本概念和应用,以及其中的一些新进展。
一、基因序列分析的基本概念基因序列是指基因在染色体上的排列顺序。
基因序列分析是指对基因序列进行分析并研究其特性和功能。
通过基因序列分析,可以发现基因的结构和功能,进而研究基因在生命活动中的作用。
基因序列分析的方法主要包括以下几种:1.基因定位:通过分析基因在染色体上的位置,确定基因的位置和数量。
2.基因标记:利用常见的DNA多态性技术,将基因与某些特定的DNA序列联系起来,便于搜寻和分析。
3.基因表达:对基因表达的研究可以揭示基因在不同组织中的表达量和差异,进一步分析基因的功能和作用。
4.序列比对:通过比对不同生物的基因序列,发现基因序列的变异情况和演化趋势。
二、生物信息学的基本概念生物信息学是指将计算机科学和生命科学相结合,研究生物大分子的结构、功能以及相互作用等问题的学科领域。
生物信息学的目标是将海量的生物信息抽象出来形成一个数据库或者算法,为人类解决生命科学问题提供基础支撑。
生物信息学的方法主要包括以下几种:1.序列分析:通过对DNA、RNA和蛋白质序列进行比对和分析,研究序列的相似性和差异性,发现与基因相关的信息。
2.结构分析:利用计算机模拟技术和X射线衍射技术,对生物分子的结构进行分析和预测,揭示分子的结构与功能之间的关系。
3.生物网络分析:通过对生物系统的建模和分析,预测生物相互作用网络的复杂关系。
三、应用案例基因序列分析和生物信息学技术在生物学领域中有着广泛的应用,以下是几个典型的应用案例。
1.癌症基因剖析:先通过基因测序寻找潜在的基因突变,然后利用生物信息学的方法分析基因功能、突变的可能影响以及治疗方案。
2.基因工程:将生物信息技术技术和基因工程技术相结合,可以对基因进行修改和改造,实现人造生物的创造和生产。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
Gene 序列分析原文/vionit/blog/item/98edb0dc706167a2cc116651.html核酸和蛋白质序列分析在获得一个基因序列后,需要对其进行生物信息学分析,从中尽量发掘信息,从而指导进一步的实验研究。
通过染色体定位分析、内含子/外显子分析、ORF分析、表达谱分析等,能够阐明基因的基本信息。
通过启动子预测、CpG岛分析和转录因子分析等,识别调控区的顺式作用元件,可以为基因的调控研究提供基础。
通过蛋白质基本性质分析,疏水性分析,跨膜区预测,信号肽预测,亚细胞定位预测,抗原性位点预测,可以对基因编码蛋白的性质作出初步判断和预测。
尤其通过疏水性分析和跨膜区预测可以预测基因是否为膜蛋白,这对确定实验研究方向有重要的参考意义。
此外,通过相似性搜索、功能位点分析、结构分析、查询基因表达谱聚簇数据库、基因敲除数据库、基因组上下游邻居等,尽量挖掘网络数据库中的信息,可以对基因功能作出推论。
上述技术路线可为其它类似分子的生物信息学分析提供借鉴。
本路线图及推荐网址已建立超级链接,放在北京大学人类疾病基因研究中心网站(/science/bioinfomatics.htm),可以直接点击进入检索网站。
下面介绍其中一些基本分析。
值得注意的是,在对序列进行分析时,首先应当明确序列的性质是mRNA序列还是基因组序列?是计算机拼接得到还是经过PCR扩增测序得到?是原核生物还是真核生物?这些决定了分析方法的选择和分析结果的解释。
(一)核酸序列分析1、双序列比对(pairwise alignment)双序列比对是指比较两条序列的相似性和寻找相似碱基及氨基酸的对应位置,它是用计算机进行序列分析的强大工具,分为全局比对和局部比对两类,各以Needleman-Wunsch算法和Smith-Waterman算法为代表。
由于这些算法都是启发式(heuristic)的算法,因此并没有最优值。
根据比对的需要,选用适当的比对工具,在比对时适当调整空格罚分(gap penalty)和空格延伸罚分(gap extension penalty),以获得更优的比对。
除了利用BLAST、FASTA等局部比对工具进行序列对数据库的搜索外我们还推荐使用EMBOSS软件包中的Needle软件(http://bioinfo.pbi.nrc.ca:8090/EMBOSS/),和Pairwise BLAST (/BLAST/)。
以上介绍的这些双序列比对工具的使用都比较简单,一般输入所比较的序列即可。
(1)BLAST和FASTAFASTA(/fasta33/)和BLAST(/BLAST/)是目前运用较为广泛的相似性搜索工具。
这两个工具都采用局部比对的方法,选择计分矩阵对序列计分,通过分值的大小和统计学显著性分析确定有意义的局部比对。
使用FASTA和BLAST,进行数据库搜索,找到与查询序列有一定相似性的序列。
一般认为,如果蛋白的序列一致性为25-30%,则可认为序列同源。
BLAST根据搜索序列和数据库的不同类型分为5种(表2),另外PSI-BLAST通过迭代搜索,可以搜索到与查询序列相似性较低的序列。
其中BLASTN、BLASTP在实践中最为常用,TBLASTN 在搜索相似序列进行新基因预测时特别有用。
使用BLAST时,先选择需要使用的BLAST程序,然后提供相应的查询序列,选择所比对的数据库即可。
(2)Needle和Pairwise BLAST:其中Needle适用于蛋白质和DNA序列,而Pairwise BLAST仅适用于DNA序列(3)相似性和同源性:必须指出,相似性(similarity)和同源性( homology)是两个完全不同的概念。
同源序列是指从某一共同祖先经过趋异进化而形成的不同序列。
相似性是指序列比对过程中检测序列和目标序列之间相同碱基或氨基酸残基序列所占比例的大小。
经过比对,当相似性高于一定程度,可以推测序列可能是同源序列,具有一定同源性。
2、多序列比对和进化树在研究生物问题时,常常需要同时对两个以上的序列进行比对,这就是多序列比对。
多序列比对可用于研究一组相关基因或蛋白,推断基因的进化关系,还可用于发现一组功能或结构相关基因之间的共有模式(pattern)。
最常用的多序列比对工具为ClustalW (/clustalw/),多用于比较蛋白序列。
ClustalW用法:(1)输入:序列以FastA格式输入。
(2)输出:除了以文本形式外,还可以通过JalView显示和编辑结果。
此外,还可以另外使用GeneDoc(常见于文献)及DNAStar软件等显示结果。
多序列比对的结果还用于进一步绘制进化树。
3、ORF(Open Reading Frame)分析从核酸序列翻译得到蛋白质序列,需要进行ORF分析,每个生物信息学分析软件包几乎都带有翻译功能。
推荐使用NCBI的ORF Finder(/gorf/gorf.html)软件或EMBOSS中的getorf(http://bioinfo.pbi.nrc.ca:8090/EMBOSS/)软件。
ORF Finder 以图形方式,分为正链+1、+2、+3和反链+1、+2、+3六个相位预测ORF;Getorf可指定预测ORF的长度下限和指定预测正反链。
进行ORF分析虽然比较简单,但应注意以下几点:(1)序列的准确性:尤其是通过计算机拼接的序列,需要根据EST和基因组序列进行反复校正。
(2)ORF是否完整:看在ORF上游同一相位是否具有终止码,或者具有起始密码子。
(3)参考Kozak一致性规律,即起始密码子位点符合A/GCCATGG。
(4)不要忽略反义读框。
4、染色体定位根据基因组图谱对序列进行染色体定位和浏览其基因组上下游基因。
具体方法为:(1)进行Genomic BLAST搜索。
(2)通过“Genome view”观察基因组结构。
(3)点击相应染色体区域,通过表意图(ideogram)和相应区域上下游的基因进行精确定位。
5、基因结构分析根据基因的mRNA序列及基因组序列,可以进行基因结构的分析。
推荐使用BLAST或BLAT(/cgi-bin/hgBlat?command=start)进行分析。
由于真核生物转录后内含子将被剪切,因此将mRNA和基因组进行比对以后,会发现mRNA 的每个外显子与基因组序列片断匹配,根据这些片段可以判断外显子的数目和大小。
外显子和内含子具体边界的确定,可以参考GT/AG一致性规则。
BLAT的结果直接显示外显子数目、大小及边界。
6、基因上游调控区分析(1)启动子预测:推荐使用冷泉港开发的FIRSTEF程序(/tools/FirstEF/)进行启动子预测。
用RT-PCR等实验方法获得的mRNA往往缺少完整的5’端,采用FirstEF程序可以对第一外显子(尤其是非编码的第一外显子)和CpG相关启动子进行预测。
方法:以FastA格式输入起始密码子上游序列。
(2)转录因子结合位点分析:推荐使用TFSEARCH 程序(http://www.cbrc.jp/research/db/TFSEARCH.html)及MATCH程序(/pub/programs.html#match)对转录因子数据库TRANSFAC (http://transfac.gbf.de/TRANSFAC/)进行搜索,寻找可能的转录因子结合位点。
方法:输入起始密码子上游序列。
结果将给出很多可能的转录因子结合位点,注意选择其中分值较高的位点。
(二)蛋白质序列分析1、跨膜区预测各个物种的膜蛋白的比例差别不大,约四分之一的人类已知蛋白为膜蛋白。
由于膜蛋白不溶于水,分离纯化困难,不容易生长晶体,很难确定其结构。
因此,对膜蛋白的跨膜螺旋进行预测是生物信息学的重要应用。
推荐使用TMHMM软件(http://www.cbs.dtu.dk/services/TMHMM/)对蛋白进行跨膜预测。
TMHMM综合了跨膜区疏水性、电荷偏倚、螺旋长度和膜蛋白拓扑学限制等性质,采用隐马氏模型(Hidden Markov Models),对跨膜区及膜内外区进行整体的预测。
TMHMM是目前最好的进行跨膜区预测的软件,它尤其长于区分可溶性蛋白和膜蛋白,因此首选它来判定一个蛋白是否为膜蛋白。
所有跨膜区预测软件的准确性都不超过52%,但86%的跨膜区可以通过不同的软件进行正确预测。
因此,综合分析不同的软件预测结果和疏水性图以获得更好的预测结果。
方法:输入待分析的蛋白序列即可。
2、信号肽预测信号肽位于分泌蛋白的N端,当蛋白跨膜转移位置时被切掉。
信号肽的特征是包括一个正电荷区域、一个疏水性区域和不带电荷但具有极性的区域。
信号肽切割位点的-3和-1位为小而中性氨基酸。
推荐使用SignalP软件2.0版(http://www.cbs.dtu.dk/services/SignalP-2.0/)对PDCD5N端序列进行信号肽分析。
SignalP2.0根据信号肽序列特征,采用神经网络方法或隐马氏模型方法,根据物种的不同,分别选择用真核和原核序列进行训练,对信号肽位置及切割位点进行预测。
信号肽切割位点预测用Y-score maximum来判断,对是否分泌蛋白用mean S-score来判断:如果mean S-score大于0.5,则预测为分泌蛋白,存在信号肽,但II型跨膜蛋白的N端序列可能被错误预测为分泌蛋白的信号肽。
方法:输入待分析的蛋白序列,如为原核基因选择原核训练集,否则选择真核训练集。
3、亚细胞定位预测亚细胞定位与蛋白质的功能存在着非常重要的联系。
亚细胞定位预测基于如下原理:(1)不同的细胞器往往具有不同的理化环境它根据蛋白质的结构及表面理化特征,选择性容纳蛋白。
(2)蛋白质表面直接暴露于细胞器环境中它由序列折叠过程决定,而后者取决于氨基酸组成。
因此可以通过氨基酸组成进行亚细胞定位的预测。
推荐使用PSORT (http://psort.nibb.ac.jp/)II软件对PDCD5蛋白的细胞内定位进行预测。
PSORT将动物蛋白质定位于10个细胞器:(1)细胞浆,(2)细胞骨架,(3)内质网,(4)胞外,(5)高尔基体,(6)溶酶体,(7)线粒体,(8)胞核,(9)过氧化物酶体(peroxisome)和(10)细胞膜。