现代有机合成第6章
合集下载
有机合成化学6—反合成分析法

第六章逆合成分析
有机合成设计基础知识
有机合成:利用化学反应,将简单的有机化合物制成比较复杂的有机物的过程。
有机反应是合成的基础,路线设计是合成的关键
要做好有机合成设计,除了对有机单元反应和有机合成技术要熟练掌握外,还要有科学的逻辑思维方法
有机合成路线设计基本原则:
u廉价易得的原料、尽可能少的反应步骤、好的选择性、尽可能高的
产率、温和的反应条件和原子经济性等。
u在工业规模的合成上,尽可能地减少环境污染,采用温和的反
应条件以及易于产品分离的路线等。
Me a a
7.2 逆向切断技巧
在逆合成分析中,简化目标分子的最有效手段是切断,不同的切断方式和切断顺序都将导致不同的合成路线。
1. 优先考虑骨架的形成
2. 碳-杂键优先切断
3. 官能团部位先切断
4. 添加辅助基团后切断
5. 逆推到适当阶段再切断
6. 利用分子的对称性
O
O O
H
H
Ph。
有机合成课件6章(缩合反应)
OH
2CH3COCH3 Ba(OH)2 CH3COCH2C(CH3)2 I2 CH3COCH=C(CH3)2
第六章 缩合反应
甲醛和苯甲醛不含α-H,可以和含活泼α-H的醛或酮缩合:
O
HC-O + CH3CHO
[HOCH2CH2CHO]
CH2=CH-CHO
Ph CH=O + CH3COCH3
HO Ph-C=C-C-CH3
NaOC2H5 NaOC2H5
PhCH CO2C2H5 CHO
PhCH(CO2C2H5)2 86%
如果把醛或酮滴加入不含活泼α-氢的酯溶液中,由于酯本身 不能自身缩合,而醛(酮)本身浓度很小,自身缩合的机会很 小,此时仍能得到较高收率的缩合产物。
第六章 缩合反应
如: PhCO2C2H5 + CH3COPh
O
PhCOCl H2N CH2CO2H
PhCONH CH2CO2H -H2O
O
N
PhCHO
Ph CH
O O
还原
H2 Ph C
O O
Ph
CH3CO2K
N Ph
Ph CH2 C CO2H NH
PhCH
O H2O Ph CH2 C CO2H
N
Ph
H2O C CO2H
H2O
NH2
PhCH2CH CO2H
NH COPh
CHO (CH3CO)2O KOCOCH3
CH=CH CO2H
这是制备肉桂酸的基本方法。能发生类似反应的还有其它醛。
(CH3CO)2O O CH O KOCOCH3
O CH=CH CO2H 65~70%
第六章 缩合反应
O2O2N 80%
有机化学第六章芳香烃

Y
可见,凯库勒式并不能确切地反映苯的真实情况
现代物理方法(射线法、光谱法、偶极距的测定)表明,苯分子是 一个平面正六边形构型,键角都是120°,碳碳键长都是0.1397nm。图 示如下:
杂化轨道理论解释
苯分子中的碳原子都是以sp2杂化轨道互相沿对称轴方向重叠形成6个C-Cσ键组成一个 正六边形,每个C各以一个sp2杂化轨道分别与H的1s轨道沿对称的方向重叠,形成六 个C-Hσ键,由于是sp2杂化,所以键角都是120。所有原子均在同一平面上。 每个C还有一个未参与杂化的垂直于与碳环平面σ键的P轨道,彼此侧面重叠,形成一 个封闭的共轭体系,每个P轨道上有一个P电子,组成了π66大π键。由于共轭效应使π 电子高度离域,电子云完全平均化,故无单双键之分。 因此,苯的电子云是一个整体,分布在环的上、下方,并且是完全平均的,所以苯分 子中每个C-C键都有π键的性质,并且是完全相同的,故邻位二元取代物也应当只有一 种。 应当注意且要牢记,苯环中并没有一般的C-C单键和C=C双键。
( 2 )体系能量降低,氢化热(208.5 kJ·mol-1)比环己烯氢 化热的三倍低得多( 3×119.3-208.5 = 149.4 kj·mol-1 ),这 149.4 kj·mol-1即为苯的共轭能。
苯现在的表达方式
价键式
分子轨道离域式
共振式
自旋偶合价键理论 (1986年Copper等提出)
+ Cl2 + Br2
Fe 或 FeCl3 55~60℃
Fe 或 FeBr3 55~60℃
+ 2Cl2 Fe 或 FeCl3
反应历程:
Cl
+ HCl
Br
+ HBr
Cl
+
有机合成课件6章(缩合反应)(最新版)
NH
CH CH Ph
H3C O2N P2N O NO2
4. Knoevenagel反应: 这类反应的特点是一个亚甲基上连接两个吸电子基团, 使得其氢活性明显提高,反应较易进行。一般使用弱碱(有 机胺)作催化剂即可,甚至不使用催化剂。
第六章 缩合反应
如:
CH2(CN)2
O CH2-C-H
CH3CHO
OH CH3-CH-CH2-CH=O 不稳定,易脱水
CH3CH=CH-CHO
从反应过程来看,应该有两种可能缩合产物。表示如下:
第六章 缩合反应
O CH2-C-H (1) CH3-CH=O CH2=CH-O O H C-CH3 (2) CH3-CH-CH2-CH=O O CH3-CH-CH2-CHO (1) OH H =-16.62KJ/mol
和卤代酸酯的反应(Darzenes) 该反应也要在强作用下完成。如:
O + ClCH2CO2C2H5 t BuOK 83~95% O CO2C2H5
其反应过程为:
ClCH2CO2C2H5 Cl
t BuOK
ClCHCO2C2H5
O
CHCO2C2H5 O O
CO2C2H5
第六章 缩合反应
3,醛(酮)与其它化合物的缩合: 1)硝基化合物:
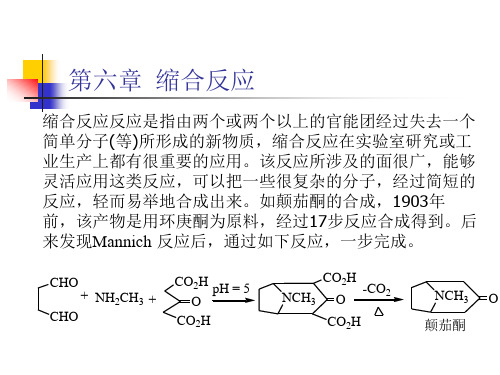
CHO CHO + NH2CH3 + CO2H O CO2H pH = 5 CO2H NCH3 O CO2H -CO2 NCH3 O
颠茄酮
第六章 缩合反应
一,碳负离子历程的缩合反应
这类反应很多,如羟醛(酮) 缩合,酯缩合等。其反应历程为:
C + C= O C O C 产物
1,醇醛缩合:
CH3CHO NaOH -H2O
CH CH Ph
H3C O2N P2N O NO2
4. Knoevenagel反应: 这类反应的特点是一个亚甲基上连接两个吸电子基团, 使得其氢活性明显提高,反应较易进行。一般使用弱碱(有 机胺)作催化剂即可,甚至不使用催化剂。
第六章 缩合反应
如:
CH2(CN)2
O CH2-C-H
CH3CHO
OH CH3-CH-CH2-CH=O 不稳定,易脱水
CH3CH=CH-CHO
从反应过程来看,应该有两种可能缩合产物。表示如下:
第六章 缩合反应
O CH2-C-H (1) CH3-CH=O CH2=CH-O O H C-CH3 (2) CH3-CH-CH2-CH=O O CH3-CH-CH2-CHO (1) OH H =-16.62KJ/mol
和卤代酸酯的反应(Darzenes) 该反应也要在强作用下完成。如:
O + ClCH2CO2C2H5 t BuOK 83~95% O CO2C2H5
其反应过程为:
ClCH2CO2C2H5 Cl
t BuOK
ClCHCO2C2H5
O
CHCO2C2H5 O O
CO2C2H5
第六章 缩合反应
3,醛(酮)与其它化合物的缩合: 1)硝基化合物:
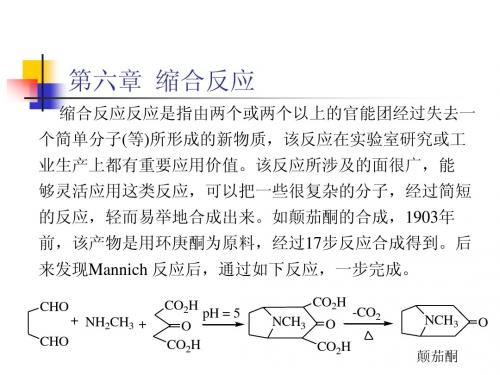
CHO CHO + NH2CH3 + CO2H O CO2H pH = 5 CO2H NCH3 O CO2H -CO2 NCH3 O
颠茄酮
第六章 缩合反应
一,碳负离子历程的缩合反应
这类反应很多,如羟醛(酮) 缩合,酯缩合等。其反应历程为:
C + C= O C O C 产物
1,醇醛缩合:
CH3CHO NaOH -H2O
有机化学第6章卤代烃12.概要

CH3CH2CH2CH2MgBr
Br + Mg THF THF MgBr 苯基溴化镁
四氢呋喃
Grignard Reagent在有机合成中应用非常广泛,是最
重要的有机金属化合物(金属原子直接与C原子连接的有 机物)之一。
R
R
O R Mg X O R R
以无水乙醚作溶剂,因为它可与格氏试剂形成路易 斯酸和路易斯碱的络合物而使格氏试剂稳定。
一般过渡态能量最高一步的反应的活化能最高反应的速率最小这一多步反应的整个反应的速率就取决于这最慢的一步即决定整个反应速率的一步叫作速率决定步骤ratedeterminingstep简rds在一个多步反应中每一步的反应速率是不同的有快bimolecularnucleophlicsubstitutionunimolecularnucleophilicsubstitution631双分子亲核取代反应s2p138bimolecularnucleophlicsubstitutionchhochbroh二级反应反应速率与溴甲烷及碱的浓度成正比
而把有Grignard试剂参与的反应,称为Grignard反应。
重点掌握
卤代烷与金属镁反应的活性顺序是:
> RF RI > RBr > RCl >
CH3CH2Br
+
RX > ArX
Mg
无水乙醚
CH3CH2MgBr 乙基溴化镁
Mg 无水乙醚
CH3CH2CH2CH2OH
HBr
CH3CH2CH2CH2Br
*卤代烷水解制醇较少应用。
2)醇解反应----被烷氧基取代 (-OR)
卤代烷与醇钠(或酚钠)作用, X 被 -OR取代, 生成醚。
R—X + NaOR’——> R—O—R’+ NaX
精细有机合成化学以及工艺学 第六章 硝化以及亚硝化

15
6.3.6硝化副反应 (1)主要的硝化副反应:氧化、去烃基、置换、 脱羧、开环、聚合等。其中氧化影响最大,生成 硝基酚。 (2)烷基苯硝化时,硝化液颜色发黑变暗,说明 硝酸用量不足或硝化温度过高。 因 为 形 成 了 络 合 物 (C6H5CH3· 2ONOSO3H· 2SO4)。 3H 破坏方法:45~55℃时补加HNO3或混酸 (3)大多数副反应,与体系中存在的氮的氧化物 有关。
-8
1.210
-8
10
结论:苯环上有给电子基,硝化反应速度快,产物 主要是邻、对位 苯环上有吸电子基,硝化速度减慢,产物主要 是间位。 2.芳烃硝化异构产物 带有吸电子的取代芳烃硝化邻位异构体 的生成量比对位异构体多。原因是硝基易同 邻位取代基中带负电荷的原子形成络合物。
CHO 19 72 9 1.3 COOH 18.5 80.5 2.2 CN 17.1 80.7 1 NO 2 9 90 8.2 40.8 NO 2 51 OCH 3
F . N . A. D.V .S . 1 F . N . A. D.V .S . F . N . A. 100 % 1 D.V .S .
22
6.4.2 混算配制 配酸工艺:水、硫酸、硝酸如何混合? 先将浓硫酸先缓慢、后渐快加入到水中, 在40度以下先慢后快加入硝酸。
23
6.4.3硝化操作
(4)非均相混酸硝化
被硝化物和硝化产物在反应温度下都呈液态,且 难溶于混酸时,常采用非均相的混酸硝化。这时 需剧烈搅拌,使有机相充分地分散到酸相中以完 成硝化反应。
(5)有机溶剂中硝化
可避免大量使用硫酸作溶剂。 (6)气相硝化 NO2与苯于80~190℃通过分子筛处理便转化为 硝基苯。
5
6.2硝化理论解释
有机化学 第六章 芳香烃
第六 章 芳烃 芳香性
(一) 芳烃的构造异构和命名 (二) 苯的结构 (三) 单环芳烃的来源 (四) 单环芳烃的物理性质 (五) 单环芳烃的化学性质 (六) 苯环上取代反应的定位规则 (七) 稠环芳烃 (八) 芳香性 (九) 富勒烯
第六章 芳烃 芳香性
• 芳烃——芳香族碳氢化合物。含有苯环的一 大类C、H化合物。 “芳香”二字的含义:
1,2,4,5-四甲苯
(2) 命名
命名时,一般以芳环为取代基,也可以芳环为母体。具
体情况,具体对待:
CH=CH2
CH=CH2
苯乙烯
对二乙烯基苯 CH=CH2
CH2Cl
CH2OH
苯氯甲烷 氯苄
苯甲醇 苄醇
• C6H5- 苯基(Ph-) ;
C6H5CH2- 苄基 ;
Ar- 芳基(芳环上去掉一个氢后,所剩下的原子团);
O
慢
H
SO3-
快 HSO4-
+
σ-络合物
SO3- 快
H3O+
SO3H + H2O
(丁) 烷基化反应机理
苯环烷基化反应中,AlCl3的作用是与卤烷起反应, 加速R+的生成:
RCl + AlCl3
R+ + AlCl4-
亲电试剂
+ R+
R
+H
σ-络合物
AlCl4-
R + HCl + AlCl3
苯环烷基化反应时,产生异构化的原因:
Br
p-二溴苯
注意:第二个卤素原子进入第一个卤素原子的邻、对位。
(乙) 硝化
+ HNO3
浓H2SO。4
50-60 C
(一) 芳烃的构造异构和命名 (二) 苯的结构 (三) 单环芳烃的来源 (四) 单环芳烃的物理性质 (五) 单环芳烃的化学性质 (六) 苯环上取代反应的定位规则 (七) 稠环芳烃 (八) 芳香性 (九) 富勒烯
第六章 芳烃 芳香性
• 芳烃——芳香族碳氢化合物。含有苯环的一 大类C、H化合物。 “芳香”二字的含义:
1,2,4,5-四甲苯
(2) 命名
命名时,一般以芳环为取代基,也可以芳环为母体。具
体情况,具体对待:
CH=CH2
CH=CH2
苯乙烯
对二乙烯基苯 CH=CH2
CH2Cl
CH2OH
苯氯甲烷 氯苄
苯甲醇 苄醇
• C6H5- 苯基(Ph-) ;
C6H5CH2- 苄基 ;
Ar- 芳基(芳环上去掉一个氢后,所剩下的原子团);
O
慢
H
SO3-
快 HSO4-
+
σ-络合物
SO3- 快
H3O+
SO3H + H2O
(丁) 烷基化反应机理
苯环烷基化反应中,AlCl3的作用是与卤烷起反应, 加速R+的生成:
RCl + AlCl3
R+ + AlCl4-
亲电试剂
+ R+
R
+H
σ-络合物
AlCl4-
R + HCl + AlCl3
苯环烷基化反应时,产生异构化的原因:
Br
p-二溴苯
注意:第二个卤素原子进入第一个卤素原子的邻、对位。
(乙) 硝化
+ HNO3
浓H2SO。4
50-60 C
有机化学 第六章 卤代烃
+
+
H2 O ( C 2 H5 O H)
( C 2 H 5 O -)
第六章 卤代烃
36 15:49
单分子历程(E1)(续)
OH-、C2H5O-作为亲核试剂与碳正离子结合,生成醇或醚
R1 HOR C H2 R1 R2 R C H2 C+ R2 C 2H5OR C H2 C R2 O C 2H5 醚 R1 C OH 醇
E2表示。
第六章 卤代烃
34 15:49
单分子历程(E1)
首先生成碳正离子中间体:
R1 慢 R C H2 C R2 X R C H2 C+ R2 R1
+
X
下一步反应可能有两种情况: 第六章 卤代烃
35 15:49
单分子历程(E1)(续)
OH-、C2H5O-作为碱由-碳原子上夺取一个氢,生成烯
H HOR1 快 RCH C+ R2 RCH C R2 R1
9 15:49
亲核取代反应:起始于亲核试剂的进攻而发生的取代 反应(99页)※
Nu:-
+
+
R C
X R C : Nu
+
:X
亲核试剂
底物
离去基团
※三个概念:亲核试剂、底物、离去基团(99页) 第六章 卤代烃
10 15:49
①:被羟基取代:NaOH或KOH水溶液中共热, 生成醇。该反应被称为卤代烃的水解。
21 15:49
在化学动力学中,反应速率决定于反应中最慢的一步,反
应分子数则由决定反应速率的一步来衡量。上述历程
中第一步是决定反应速率的一步,而这一步决定于C-X
键的断裂,与作用试剂无关,所以叫做单分子历程。
有机化学 06第六章 卤代烃2
离去基团的影响:
R-Cl
R-Br
R-I
反应速度增大
6.3.2 消除反应 E (Elimination reaction)
βα
醇
R CH CH 2 + NaOH △
HX
RCH=CH 2 + NaX + H 2O
从分子中脱去一个简单小分子,如HX、H2O等,同时 产生不饱和键的反应称为消除反应。
反应中除α碳脱去X外,在β碳上脱去H,故称为β-消 除反应。
C2H5O- + CH3
CH3 C CH3
Br
[C2H5O-
进攻-H
] H
CH3
CH2 C CH3
Br
CH 3 CH3 C =CH2 + C2H5OH + Br-
SN2反应机理
HO- + CH3Br
[ ] H H HO C Br
进攻-C H
CH3OH + Br-
试剂碱性强,升高温度有利于E2反应。
四、亲核取代反应与消除反应的关系
醇溶液
胺RNH2 + HX
RONO2 + Ag X
硝酸酯
亲核取代反应通式:
RCδ+H2 Xδ- + Nu -
RCH2Nu + X -
反应底物
亲核试剂
产物
离去基团
卤代烷
HO- 、CN- 、 OR-、NH3 ONO-2等
醇、腈、 醚、胺 硝酸酯等
卤素离子
由试剂的负离子部分或未共用电子对去进攻而引
发反应,进攻试剂都有较大的电子云密度,能提供一
写出下列反应的主要产物
CH3
Br NaOH ,C2H5OH
有机合成第6章有机合成中的选择性
醚化保护
将醇官能团转化为醚,以防止其在反应中的氧化 或脱水。常用的醚化试剂包括醇和烷基卤化物。
酰化保护
将胺官能团转化为酰胺,以避免其在反应中的碱 性干扰。常用的酰化试剂包括酰氯、酸酐或酯。
官能团去保护方法
01
02
03
酯的水解
在酸性或碱性条件下,酯 可以水解回羧酸。酸性水 解通常使用稀酸,而碱性 水解则使用稀碱。
PART 05
化学选择性合成策略
REPORTING
WENKU DESIGN
底物设计策略
选择性底物设计
通过改变底物的结构或官能团,实现目标产物的选择 性合成。
底物活性调控
通过改变底物的电子效应、空间效应或反应中间体的 稳定性,提高目标产物的选择性。
底物保护策略
采用保护基团对底物中的敏感官能团进行保护,避免 不必要的副反应,提高目标产物的选择性。
底物与催化剂的匹配
选择合适的底物和催化剂,使它们之间形成具有特定空间排列的过 渡态,从而实现立体选择性控制。
反应条件的优化
通过调整反应温度、压力、溶剂等条件,优化不对称催化反应的效 果,提高目标产物的立体选择性。
手性拆分法
消旋体的制备
首先制备外消旋体或内消旋体,作为手性拆分的起始原料。
手性拆分剂的选择
手性传递
通过化学反应将手性源的立体化学 信息传递给目标分子,保持或增强 手性纯度。
手性放大
利用手性催化剂或手性试剂将手性 源的微量不对称性放大,实现目标 分子的高立体选择性合成。
不对称催化反应法
不对称催化剂的设计与合成
设计并合成具有特定空间结构和活性的不对称催化剂,以实现目 标反应的高立体选择性。
反应温度控制
通过控制反应温度,改变反应速率和 选择性,实现目标产物的选择性合成。
将醇官能团转化为醚,以防止其在反应中的氧化 或脱水。常用的醚化试剂包括醇和烷基卤化物。
酰化保护
将胺官能团转化为酰胺,以避免其在反应中的碱 性干扰。常用的酰化试剂包括酰氯、酸酐或酯。
官能团去保护方法
01
02
03
酯的水解
在酸性或碱性条件下,酯 可以水解回羧酸。酸性水 解通常使用稀酸,而碱性 水解则使用稀碱。
PART 05
化学选择性合成策略
REPORTING
WENKU DESIGN
底物设计策略
选择性底物设计
通过改变底物的结构或官能团,实现目标产物的选择 性合成。
底物活性调控
通过改变底物的电子效应、空间效应或反应中间体的 稳定性,提高目标产物的选择性。
底物保护策略
采用保护基团对底物中的敏感官能团进行保护,避免 不必要的副反应,提高目标产物的选择性。
底物与催化剂的匹配
选择合适的底物和催化剂,使它们之间形成具有特定空间排列的过 渡态,从而实现立体选择性控制。
反应条件的优化
通过调整反应温度、压力、溶剂等条件,优化不对称催化反应的效 果,提高目标产物的立体选择性。
手性拆分法
消旋体的制备
首先制备外消旋体或内消旋体,作为手性拆分的起始原料。
手性拆分剂的选择
手性传递
通过化学反应将手性源的立体化学 信息传递给目标分子,保持或增强 手性纯度。
手性放大
利用手性催化剂或手性试剂将手性 源的微量不对称性放大,实现目标 分子的高立体选择性合成。
不对称催化反应法
不对称催化剂的设计与合成
设计并合成具有特定空间结构和活性的不对称催化剂,以实现目 标反应的高立体选择性。
反应温度控制
通过控制反应温度,改变反应速率和 选择性,实现目标产物的选择性合成。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
现代有机合成化学
第六章 有机合成概论
鉴于目前文献上合成元与合成中间体的混淆使用,Corey在反合成转 化方面又提出了一个术语,即反合成元。反合成元意为进行某一转化 所必要的结构单元。如下列结构单元A,B,C,D分别为Diels-Alder 反应、Claisen重排、Robinson造环、Mannich转化的基本反合成元。
现代有机合成化学
第六章 有机合成概论
现代有机合成化学
第六章 有机合成概论
其他的天然资源,如糖中最廉价的葡萄糖一直也是常常被使用于合 成研究的出发原料,例如用于白三烯A4的合成。
现代有机合成化学
第六章 有机合成概论
氨基酸中最廉价的是L—谷氨酸(L-Glutamic Acid),它的利用也是合成设计经常 考虑的手性原料(参阅G.M.Coppola et al.Asymmetric Synthesis, John Wiley & Son,NY,1987,p.216.)
目标分子中官能团的反合成转化也像合成反应中一样有三种类型:变换
。 (interconversion) (FGl),引入 (addition) (FGA)和消除(removal)
(FRG)
现代有机合成化学
第六章 有机合成概论
重复或交替使用上述各转化过程也就可以推导出合成目标分子所 需的易得的原料,如:
试剂原料,类似这 样推导出的图像如 向下长的树,所以 也称合成树。实际 工作中不可能将每 一条路线都去实验 室实践,所以还必 须对合成树进行修 剪,也就是必须考 察、比较并进行取 舍。
现代有机合成化学
第六章 有机合成概论
目录
6.1
有机合成的三种出发点
6.2
合成设计的三部曲
现代有机合成化学
第六章 有机合成概论
有机合成是有机化学中一个古老的分支,也是一个十分活跃 的区域,有机化学家为了基础理论和应用研究的需要,不断从事 着已知或未知结构有机分子的合成,今天我们将它称为有机分子 工程学。由Corey提出并由此发展起来的“合成元”(synthon)、 “反合成分析”(retrosynthesis或antisynthesis)、“反合成 元”(retron)的概念是当今有机合成中最为普遍接受的设计方法 论.Corey的反合成分析被有机合成化学界称作是Harvard学派的代 表,与Cambridge学派的生源合成学说一起组成了现代有机合成设 计思想的基座.本章及第七、八章就从Corey的这些基本概念出发, 对有机合成设计的方法作一概性的介绍.
由于其中只有a-异构体能引诱甲虫,所以必须考虑另外能控制立体化学的 反合成转化,这些比较新的考虑我们将在后面提及。
除立体化学问题外,诸如合成的经济性问题,包括路线长短,产率,原料 易得性,分离方法等都必须加以考察,这样才能筛选出最佳的合成设计。
现代有机合成化学
第六章 有机合成概论
联接和重排这两类转化通常在双线箭头上加注,这时的合成元通常即为 试剂,中间体,无需进一步推导,如下表的例子:
现代有机合成化学
第六章 有机合成概论
6.1 有机合成的三种出发点
有机化学合成实验室从事的研究,其出发点不外乎下列三种:将新 发现的或有趣的反应贯彻于有意义的合成工作中;利用天然界或生 产中未充分利用的原料来合成有价值的产品;以及合成因某种需要 而提出的特定目标分子。
6.1.1 利用新反应 一些反应机理或合成方法学的研究者发现了一些新的有趣的反应,
现代有机合成化学
第六章 有机合成概论
第三步是从合成方向上进行检查,也就是对合成树的剪裁、取舍。有机合 成的工作内容概而言之可以说有三个任务:碳骨架的建立、官能团的配置以 及确立正确的立体化学。前两个任务在上一步设计中已经提及,现在的这一 步则着重要考察立体化学的问题.如欧洲榆树甲虫的集合信息素a-multistratin 有四个手性中心,如仅按下式所示的反合成设计获得的合成路线,最后将会 得到立体异构体的混合物。
现代有机合成化学
第六章 有机合成概论
Overman等发现2-烯基-1,2-二醇体系在酸存在下与醛发生缩合反应时会 发生重排而扩环,反应具有很好的立体选择性.由此,他们设计合成了 一系列天然产物.。
现代有机合成化学
第六章 有机合成概论
6.1.2 利用原料的合成
某些来源丰富的天然资源或生产中未被利用的副产物、边角料常 给有机合成化学家提出很有意义的研究课题,即如何利用它们作为原 料合成出更高价值的产品,目前这些工作被纳入资源化学的研究范 围.如α-pinene是我国松节油中的主要成分,如何利用以合成诸如芳 樟醇、龙脑之类的香料或先保留环丁环再进一步利用,这已经成为一 些合成课题组的研究课题。
以原料为出发点的合成设计,关键在于如何充分利用这一原料的结构特征和化 学反应特性。
现代有机合成化学
第六章 有机合成概论
6.1.3 特定目标分子的合成
这是在有机合成设计时最经常遇到的课题,我们将在下面的内容中 就这个议题进行深入的讨论。另外,如我们仔细考察以上两类合成设计 时可以看到,三种出发点虽有不同,但最终还是要归结到一个特定的目 标分子,因此目标分子为出发点的合成设计原则在三类设计中都是需要 参照的。
2019/11/2
现代有机合成化学
第六章 有机合成概论
角鲨烯
O HO
COOH OH 前 列 腺 素 E2
现代有机合成化学
第六章 有机合成概论
第二步是以上述分析为基础,进而一步一步倒推出合成此目标化合 物的各种路线和可能的易得起始原料,这也就是所谓反合成 (retrosynthesis或antisynthesis),此分析思路与真正的合成正好相 反.合成中间使用各种各样的反应来形成分子的骨架,改变分子骨架上 的官能团,从而最终获得目标分子;在反合成中则是利用一系列所谓的 “ 转 化 (transformation)” 来 推 导 出 一 系 列 中 间 体 和 合 适 的 起 始 原 料.转化用双线箭头表示( )线箭头表示的反应(→).由相应的已知 或可靠的反应而进行转化所得的结构单元称之为“合成元(synthon)”, 由合成元再可以推导(用虚线表示)得相应的试剂或中间体,有时合成元 本身即是试剂或中间体.
现代有机合成化学
第六章 有机合成概论
接受电子的(a)的合 成元;给电子的(d)合成 元;自由基(r)合成元; 双电子中性的(e)合成元; 下面的表中我们可以从一些 实例看出分拆的情况:
现代有机合成化学
第六章 有机合成概论
联接和重排这两类转化通常在双线箭头上加注,这时的合成元通常即为 试剂,中间体,无需进一步推导,如下表的例子:
O
HO
COOCH3 FGI H
COOCH3 con
FGI
OCH3
O
现代有机合成化学
第六章 有机合成概论
还需要指出的是, 除了最简单的目标以外, 都会有不止一条的反合 成路线分子愈复杂这种 可能的反合成路线就越 多,棉红铃虫性信息素 是一个16碳原子直链醇 的乙酸酯,结构并不算 复杂,但其反合成的可 能途径则很多。下面是 根据已报道成功合成的 路线,而画出的反成途 径合成图中仅表明碳骨 架的分拆和最后推出。
现代有机合成化学
第六章 有机合成概论
6.2 合成设计的三部曲
考虑对一个特定目标分子的合成,第一步是对这个分子的结构特 征和已知的理化性质进行收集和考察,由此可以简化合成中的问题或 者避免不必要的弯路。诸如角鲨烯的合成,30个碳的三萜分子是一个 中心对称的化合物,就可以设计一条路线,从中间出发两边对称地同 时进行合成;而考虑前列腺素E2的合成时,由于已知分子中β -羟基酮 体系是很不稳定的,因此就可以安排在合成路线的最后几步时再形成 这一结构单元,使其避免经历较多的化学反应。
现代有机合成化学
第六章 有机合成概论
吴毓林等在白三烯合成中发现下列双羟基化反应选择性较好,进而就 设计了利用此反应来合成蚊子产卵地的信息素
现代有机合成化学
第六章 有机合成概论
黄耀曾等发现砷Ylide合成共轭的醛、酮、酰胺条件平和,产率好, 进而也设计合成了一些多烯型天然产物,如:
陆熙炎等]发现了一种有效合成双环[3.3.1]壬-9-酮体系的新方法, 而许多天然产物,如石杉碱(huperzine)等就含有这一类骨架,因此 可设计利用这一反应以合成这一类天然产物及其类似物.这一方法后 为美国化学家所采用并实践于huperzine A的合成中
A
O B
O C
O N
D
现代有机合成化学
第六章 有机合成概论
有时目标分子中仅包含部分反合成元,如下列E,F,G即含有 Robinson造环的部分反合成元,它们通过若干转化就可以得到完整的 反合成元。
O
O
反合成分析的核E心问题是转化,反合F 成元和合成元则是G这一问题的两 个方面:前者是转化的必要结构单元,后者是转化将得到的结构单元。 转化同样有两大类型,即碳一碳键的转化和官能团的转化。目标分子 碳—碳骨架的转化包括分拆(disconnection),联接(connection)和 重排(rearrangement)三种。
OHOBiblioteka COOCH3 FGI HCOOCH3 con
FGI
OCH3
O
较简单的目标分子经几步转化就可以得到合成的起始原料,但较复杂的
目标分子则就需要长得多的转化,如不算最复杂的维生素A的反合成,下
面是一个合成的途径,图中为简化起见,不写出合成元,而直接写出相应
于该合成元的中间体或试剂。
现代有机合成化学
第六章 有机合成概论
现代有机合成化学
第六章 有机合成概论
但与松节油共同产生的松香[主要成分松香酸(abietic acid)]除一些 低级用途外,还未得到很好的利用,类似的情况还有松柏烯 (cembrene)等
青蒿的生长遍布全球,迄今所知除我国南方数省所产的植物含较多 青蒿素外,其他地区所产主要含青蒿酸,因而青蒿酸的利用也就成 为一些合成工作者,特别是我国科学家的研究课题。
第六章 有机合成概论
鉴于目前文献上合成元与合成中间体的混淆使用,Corey在反合成转 化方面又提出了一个术语,即反合成元。反合成元意为进行某一转化 所必要的结构单元。如下列结构单元A,B,C,D分别为Diels-Alder 反应、Claisen重排、Robinson造环、Mannich转化的基本反合成元。
现代有机合成化学
第六章 有机合成概论
现代有机合成化学
第六章 有机合成概论
其他的天然资源,如糖中最廉价的葡萄糖一直也是常常被使用于合 成研究的出发原料,例如用于白三烯A4的合成。
现代有机合成化学
第六章 有机合成概论
氨基酸中最廉价的是L—谷氨酸(L-Glutamic Acid),它的利用也是合成设计经常 考虑的手性原料(参阅G.M.Coppola et al.Asymmetric Synthesis, John Wiley & Son,NY,1987,p.216.)
目标分子中官能团的反合成转化也像合成反应中一样有三种类型:变换
。 (interconversion) (FGl),引入 (addition) (FGA)和消除(removal)
(FRG)
现代有机合成化学
第六章 有机合成概论
重复或交替使用上述各转化过程也就可以推导出合成目标分子所 需的易得的原料,如:
试剂原料,类似这 样推导出的图像如 向下长的树,所以 也称合成树。实际 工作中不可能将每 一条路线都去实验 室实践,所以还必 须对合成树进行修 剪,也就是必须考 察、比较并进行取 舍。
现代有机合成化学
第六章 有机合成概论
目录
6.1
有机合成的三种出发点
6.2
合成设计的三部曲
现代有机合成化学
第六章 有机合成概论
有机合成是有机化学中一个古老的分支,也是一个十分活跃 的区域,有机化学家为了基础理论和应用研究的需要,不断从事 着已知或未知结构有机分子的合成,今天我们将它称为有机分子 工程学。由Corey提出并由此发展起来的“合成元”(synthon)、 “反合成分析”(retrosynthesis或antisynthesis)、“反合成 元”(retron)的概念是当今有机合成中最为普遍接受的设计方法 论.Corey的反合成分析被有机合成化学界称作是Harvard学派的代 表,与Cambridge学派的生源合成学说一起组成了现代有机合成设 计思想的基座.本章及第七、八章就从Corey的这些基本概念出发, 对有机合成设计的方法作一概性的介绍.
由于其中只有a-异构体能引诱甲虫,所以必须考虑另外能控制立体化学的 反合成转化,这些比较新的考虑我们将在后面提及。
除立体化学问题外,诸如合成的经济性问题,包括路线长短,产率,原料 易得性,分离方法等都必须加以考察,这样才能筛选出最佳的合成设计。
现代有机合成化学
第六章 有机合成概论
联接和重排这两类转化通常在双线箭头上加注,这时的合成元通常即为 试剂,中间体,无需进一步推导,如下表的例子:
现代有机合成化学
第六章 有机合成概论
6.1 有机合成的三种出发点
有机化学合成实验室从事的研究,其出发点不外乎下列三种:将新 发现的或有趣的反应贯彻于有意义的合成工作中;利用天然界或生 产中未充分利用的原料来合成有价值的产品;以及合成因某种需要 而提出的特定目标分子。
6.1.1 利用新反应 一些反应机理或合成方法学的研究者发现了一些新的有趣的反应,
现代有机合成化学
第六章 有机合成概论
第三步是从合成方向上进行检查,也就是对合成树的剪裁、取舍。有机合 成的工作内容概而言之可以说有三个任务:碳骨架的建立、官能团的配置以 及确立正确的立体化学。前两个任务在上一步设计中已经提及,现在的这一 步则着重要考察立体化学的问题.如欧洲榆树甲虫的集合信息素a-multistratin 有四个手性中心,如仅按下式所示的反合成设计获得的合成路线,最后将会 得到立体异构体的混合物。
现代有机合成化学
第六章 有机合成概论
Overman等发现2-烯基-1,2-二醇体系在酸存在下与醛发生缩合反应时会 发生重排而扩环,反应具有很好的立体选择性.由此,他们设计合成了 一系列天然产物.。
现代有机合成化学
第六章 有机合成概论
6.1.2 利用原料的合成
某些来源丰富的天然资源或生产中未被利用的副产物、边角料常 给有机合成化学家提出很有意义的研究课题,即如何利用它们作为原 料合成出更高价值的产品,目前这些工作被纳入资源化学的研究范 围.如α-pinene是我国松节油中的主要成分,如何利用以合成诸如芳 樟醇、龙脑之类的香料或先保留环丁环再进一步利用,这已经成为一 些合成课题组的研究课题。
以原料为出发点的合成设计,关键在于如何充分利用这一原料的结构特征和化 学反应特性。
现代有机合成化学
第六章 有机合成概论
6.1.3 特定目标分子的合成
这是在有机合成设计时最经常遇到的课题,我们将在下面的内容中 就这个议题进行深入的讨论。另外,如我们仔细考察以上两类合成设计 时可以看到,三种出发点虽有不同,但最终还是要归结到一个特定的目 标分子,因此目标分子为出发点的合成设计原则在三类设计中都是需要 参照的。
2019/11/2
现代有机合成化学
第六章 有机合成概论
角鲨烯
O HO
COOH OH 前 列 腺 素 E2
现代有机合成化学
第六章 有机合成概论
第二步是以上述分析为基础,进而一步一步倒推出合成此目标化合 物的各种路线和可能的易得起始原料,这也就是所谓反合成 (retrosynthesis或antisynthesis),此分析思路与真正的合成正好相 反.合成中间使用各种各样的反应来形成分子的骨架,改变分子骨架上 的官能团,从而最终获得目标分子;在反合成中则是利用一系列所谓的 “ 转 化 (transformation)” 来 推 导 出 一 系 列 中 间 体 和 合 适 的 起 始 原 料.转化用双线箭头表示( )线箭头表示的反应(→).由相应的已知 或可靠的反应而进行转化所得的结构单元称之为“合成元(synthon)”, 由合成元再可以推导(用虚线表示)得相应的试剂或中间体,有时合成元 本身即是试剂或中间体.
现代有机合成化学
第六章 有机合成概论
接受电子的(a)的合 成元;给电子的(d)合成 元;自由基(r)合成元; 双电子中性的(e)合成元; 下面的表中我们可以从一些 实例看出分拆的情况:
现代有机合成化学
第六章 有机合成概论
联接和重排这两类转化通常在双线箭头上加注,这时的合成元通常即为 试剂,中间体,无需进一步推导,如下表的例子:
O
HO
COOCH3 FGI H
COOCH3 con
FGI
OCH3
O
现代有机合成化学
第六章 有机合成概论
还需要指出的是, 除了最简单的目标以外, 都会有不止一条的反合 成路线分子愈复杂这种 可能的反合成路线就越 多,棉红铃虫性信息素 是一个16碳原子直链醇 的乙酸酯,结构并不算 复杂,但其反合成的可 能途径则很多。下面是 根据已报道成功合成的 路线,而画出的反成途 径合成图中仅表明碳骨 架的分拆和最后推出。
现代有机合成化学
第六章 有机合成概论
6.2 合成设计的三部曲
考虑对一个特定目标分子的合成,第一步是对这个分子的结构特 征和已知的理化性质进行收集和考察,由此可以简化合成中的问题或 者避免不必要的弯路。诸如角鲨烯的合成,30个碳的三萜分子是一个 中心对称的化合物,就可以设计一条路线,从中间出发两边对称地同 时进行合成;而考虑前列腺素E2的合成时,由于已知分子中β -羟基酮 体系是很不稳定的,因此就可以安排在合成路线的最后几步时再形成 这一结构单元,使其避免经历较多的化学反应。
现代有机合成化学
第六章 有机合成概论
吴毓林等在白三烯合成中发现下列双羟基化反应选择性较好,进而就 设计了利用此反应来合成蚊子产卵地的信息素
现代有机合成化学
第六章 有机合成概论
黄耀曾等发现砷Ylide合成共轭的醛、酮、酰胺条件平和,产率好, 进而也设计合成了一些多烯型天然产物,如:
陆熙炎等]发现了一种有效合成双环[3.3.1]壬-9-酮体系的新方法, 而许多天然产物,如石杉碱(huperzine)等就含有这一类骨架,因此 可设计利用这一反应以合成这一类天然产物及其类似物.这一方法后 为美国化学家所采用并实践于huperzine A的合成中
A
O B
O C
O N
D
现代有机合成化学
第六章 有机合成概论
有时目标分子中仅包含部分反合成元,如下列E,F,G即含有 Robinson造环的部分反合成元,它们通过若干转化就可以得到完整的 反合成元。
O
O
反合成分析的核E心问题是转化,反合F 成元和合成元则是G这一问题的两 个方面:前者是转化的必要结构单元,后者是转化将得到的结构单元。 转化同样有两大类型,即碳一碳键的转化和官能团的转化。目标分子 碳—碳骨架的转化包括分拆(disconnection),联接(connection)和 重排(rearrangement)三种。
OHOBiblioteka COOCH3 FGI HCOOCH3 con
FGI
OCH3
O
较简单的目标分子经几步转化就可以得到合成的起始原料,但较复杂的
目标分子则就需要长得多的转化,如不算最复杂的维生素A的反合成,下
面是一个合成的途径,图中为简化起见,不写出合成元,而直接写出相应
于该合成元的中间体或试剂。
现代有机合成化学
第六章 有机合成概论
现代有机合成化学
第六章 有机合成概论
但与松节油共同产生的松香[主要成分松香酸(abietic acid)]除一些 低级用途外,还未得到很好的利用,类似的情况还有松柏烯 (cembrene)等
青蒿的生长遍布全球,迄今所知除我国南方数省所产的植物含较多 青蒿素外,其他地区所产主要含青蒿酸,因而青蒿酸的利用也就成 为一些合成工作者,特别是我国科学家的研究课题。