制药工艺学实验-广谱抗菌药氯霉素的合成
氯霉素胶囊制备实验报告

一、实验目的1. 熟悉氯霉素胶囊的制备方法;2. 掌握药物胶囊制备过程中的关键技术;3. 提高药物制剂实验技能。
二、实验原理氯霉素胶囊是一种抗生素胶囊,由氯霉素药物和胶囊壳组成。
本实验采用直接填充法制备氯霉素胶囊,将氯霉素药物粉末与胶囊壳填充剂混合均匀,填充到胶囊壳中,制成氯霉素胶囊。
三、实验材料与仪器1. 材料:氯霉素原料药、明胶、滑石粉、硬脂酸镁、食用色素等;2. 仪器:胶囊灌装机、电子天平、混合器、胶囊模具、剪刀等。
四、实验步骤1. 药物制备:将氯霉素原料药过100目筛,备用;2. 胶囊壳制备:将明胶、滑石粉、硬脂酸镁等混合均匀,制成胶囊壳填充剂;3. 混合:将氯霉素药物粉末与胶囊壳填充剂按比例混合均匀;4. 灌装:将混合好的药物填充到胶囊壳中,注意填充均匀;5. 剪切:将填充好的胶囊壳沿模具剪切,使其成型;6. 包装:将成型的氯霉素胶囊进行包装,备用。
五、实验结果与分析1. 药物制备:氯霉素原料药过100目筛后,粉末细腻,便于填充;2. 胶囊壳制备:胶囊壳填充剂混合均匀,填充剂质地细腻,有利于药物填充;3. 混合:氯霉素药物粉末与胶囊壳填充剂混合均匀,填充剂在药物粉末中分布均匀;4. 灌装:填充好的胶囊壳沿模具剪切,胶囊成型良好,无破损;5. 包装:成型的氯霉素胶囊包装整齐,无污染。
六、实验讨论1. 本实验采用直接填充法制备氯霉素胶囊,操作简便,易于掌握;2. 药物与填充剂混合均匀,有利于药物在胶囊中的分布;3. 剪切后的胶囊成型良好,无破损,符合制剂要求;4. 实验过程中,应注意药物与填充剂的混合均匀,以及填充过程中的填充量控制。
七、实验结论通过本实验,我们成功制备了氯霉素胶囊。
实验结果表明,采用直接填充法制备氯霉素胶囊操作简便,易于掌握,制备的胶囊成型良好,符合制剂要求。
本实验为进一步研究氯霉素胶囊的制备工艺提供了参考。
八、实验拓展1. 研究不同填充剂对氯霉素胶囊制备的影响;2. 探讨氯霉素胶囊的稳定性及生物利用度;3. 研究氯霉素胶囊在不同给药途径中的应用。
氯霉素工艺流程图

氯霉素工艺流程图氯霉素是一种广谱抗菌药物,被广泛用于医疗、养殖和农业行业。
下面是一份氯霉素的工艺流程图:一、原料准备:1. 取得氯霉素的原料,包括醋酸钠、4-氯-3-亚硝基苯甲酸、亚硝基乙酸甲酯、重铬酸钾、氢氧化钠等。
2. 对原料进行筛查和测试,确保原料的质量和纯度。
二、反应:1. 将亚硝基乙酸甲酯添加到反应釜中,然后加入亚硝基乙酸钠溶液。
2. 在搅拌下将反应釜加热至40-45℃,维持2-4小时。
3. 加热的同时,将4-氯-3-亚硝基苯甲酸逐渐加入反应釜中。
4. 反应釜中的液体继续保持在40-45℃下搅拌2-4小时,直到化合物充分反应。
三、中和:1. 将反应得到的溶液缓慢地加入到冷却器中。
2. 将冷却后的溶液进行中和,将溶液的酸性中和为碱性。
3. 使用适量的氢氧化钠溶液来中和。
四、沉淀:1. 在中和的过程中,使用重铬酸钾作为指示剂。
2. 当溶液中的重铬酸钾颜色由红变绿时,表示中和完成。
3. 停止加入氢氧化钠溶液,溶液中析出的沉淀物即为氯霉素。
五、过滤和干燥:1. 用真空过滤将溶液中的沉淀物过滤出来。
2. 将过滤后的沉淀物洗净并进行再过滤。
3. 将过滤后的沉淀物进行干燥,得到氯霉素的固体产物。
六、粉碎和包装:1. 对干燥后的氯霉素固体进行粉碎,使其具有合适的颗粒大小。
2. 将粉碎后的氯霉素进行包装,并进行质量检测。
3. 将符合质量要求的氯霉素产品进行存储和运输。
这是一种常见的氯霉素工艺流程图,具体的细节可能因不同的厂家和工艺条件而有所不同。
在实际操作中,还需要注重安全和环境保护,合理配置设备和工艺参数,确保生产质量和效率。
制药工艺学 氯霉素

产品
4) 问题
⊙羟醛缩合工艺终点如何控制较为科学 ⊙如果产品在醇中有一定的溶解度,母液如何处理?
第三节 氯霉素合成原理与工艺
一、DL-苏型-1-对硝基苯基2-氨基-1,3-丙二醇 1、 生产原理
2、 生产工艺
3、 生产工艺流程框图
三氯化铝 异丙醇 铝 异丙醇铝 盐酸 水 中间体 还原反应 水解反应 回收异丙醇 结晶 母液 NaOH溶液 除铝 母液 NaOH溶液 醇析
氯霉素生产工艺研究
•概述 •氯霉素中间体合成原理与工艺 对硝基苯乙酮 对硝基--乙酰胺基--羟基苯丙酮 •氯霉素合成原理与工艺 •三废处理 •结论与展望
第一节
概述
一、氯霉素的性质 氯霉素(Chloramphenicol) 分子式:C11H12O5N2CL2 结构:
化学命名:D-threo-(-)-N-[ -(hydroxymethyl)-hydroxy-p-nitrophenethyl]-2,2-dichloroacetamide D-苏式-(-)-N[- (羟基甲基)- -羟基-对硝基本 乙基]-2,2-二氯乙酰胺
回收异丙醇
氢氧化铝
产品
4、 问题
1)本工艺回收的异丙醇溶液除异丙醇外,还有那些成分? 2)异丙醇与水能形成共沸,应采用何种分离方法才能得 到无水异丙醇? 3)“亚胺物”加入水解反应中为何能提高收率?
二、DL-苏型-1-对硝基苯基2-氨基-1,3-丙二醇拆分 1、 生产原理 诱导结晶法
2、 生产工艺
3) 生产工艺流程框图
溴素 溴化氢 对硝基苯乙酮 氯苯 溴化 脱溴 产品
水
吸收
4) 问题
⊙ 水与金属离子的控制 ⊙ 水吸收的真空问题 ⊙ 脱溴过程溴素、溴化氢如何处理
制药工艺学实验-广谱抗菌药氯霉素的合成
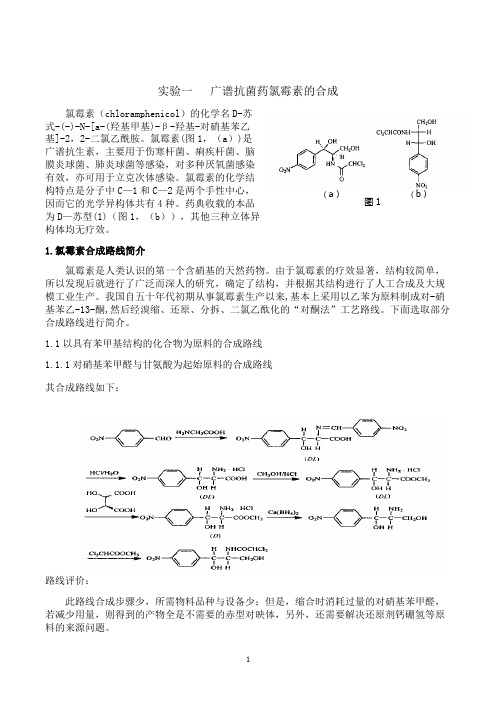
实验一 广谱抗菌药氯霉素的合成氯霉素(chloramphenicol )的化学名D-苏式-(-)-N-[a-(羟基甲基)-β-羟基-对硝基苯乙基]-2,2-二氯乙酰胺。
氯霉素(图1,(a ))是广谱抗生素,主要用于伤寒杆菌、痢疾杆菌、脑膜炎球菌、肺炎球菌等感染,对多种厌氧菌感染有效,亦可用于立克次体感染。
氯霉素的化学结构特点是分子中C —1和C —2是两个手性中心,因而它的光学异构体共有4种。
药典收载的本品为D —苏型(1)(图1,(b )),其他三种立体异构体均无疗效。
1.氯霉素合成路线简介氯霉素是人类认识的第一个含硝基的天然药物。
由于氯霉素的疗效显著,结构较简单,所以发现后就进行了广泛而深人的研究,确定了结构,并根据其结构进行了人工合成及大规模工业生产。
我国自五十年代初期从事氯霉素生产以来,基本上采用以乙苯为原料制成对-硝基苯乙-13-酮,然后经溴缩、还原、分拆、二氯乙酞化的“对酮法”工艺路线。
下面选取部分合成路线进行简介。
1.1以具有苯甲基结构的化合物为原料的合成路线1.1.1对硝基苯甲醛与甘氨酸为起始原料的合成路线其合成路线如下: 路线评价:此路线合成步骤少,所需物料品种与设备少;但是,缩合时消耗过量的对硝基苯甲醛,若减少用量,则得到的产物全是不需要的赤型对映体,另外,还需要解决还原剂钙硼氢等原料的来源问题。
图1 (a ) (b )1.1.2对硝基苯甲醛与己醛缩合经对硝基肉桂醇的合成路线对硝基苯甲醛与乙醛进行经羟醛缩合得到对硝基肉桂醛后,采用还原剂将醛还原成醇。
然后从反式对硝基肉桂醇出发经加成、环氧化、L—酒石酸铵拆分等步骤而得氯霉素。
反应路线如下:路线评价:此路线本路线使用符合立体构型要求的反式对硝基肉桂醇为中间体合成步骤不多,各步收率不低是一条有发展前途的合成路线。
1.1.3以苯甲醛为起始原料的合成路线苯甲醛与乙醛反应后再经还原得到肉桂醇,然后经下列过程制得氯霉素。
反应路线如下:路线评价:这条路线的优点是由于最后引入硝基,使得硝化反应中对位体收率高;缺点是,需要在低温下进行,需要制冷设备。
氯霉素的合成工艺

氯霉素的合成工艺11.1 概述氯霉素(Chloramphenicol),化学名为D-苏*-(-)-N-((alpha-羟甲基)-beta-羟基-beta-对硝基苯乙基)-2,2-二氯乙酰胺(D-threo-(-)-N-((alpha-hydroxymethyl)-beta-hydroxy-beta-p- nitrophenethyl)-2,2-dichloroacetamide)。
*注:在Fischer投影式中,两个相邻的手性碳原子上如有相同的原子或基团,它们不在同一边的称为苏式,在同一边的称为赤式(邢其毅:基础有机化学,第二版(上),p173)。
氯霉素为白色或微带黄绿色的针状、长片状结晶或结晶性粉末。
味苦。
熔点149~153℃。
易溶于甲醇、乙醇和丙酮等有机溶剂,微溶于水。
比旋度[alpha]D25=+18.5~+2l.5度(无水乙醇)。
氯毒素是广谱抗菌素,主要用于伤寒杆菌,痢疾杆菌、脑膜炎球菌、肺炎球菌的感染,亦可用于立克次体感染。
其主要副作用是抑制骨髓造血机能,引起粗细胞及血小板减少症或再生障碍性贫血。
但仍是治疗伤寒的首选药物。
11.2 氯霉素的合成路线氯霉素的碳骨架具苯丙基结构,按碳骨架的构建方法,氯霉素主要有两类合成路线,即分别以具有苯甲基结构和苯乙基结构的化合物为原料的合成路线。
氯霉素分子含两个手性中心,可以考虑用以下方法解决:①使用含指定手性中心的原料;②利用空间效应;③利用立体选择性的反应方法。
11.2.1 以具苯甲基结构的化合物为原料(1) 以硝基苯甲醛为原料①与甘氨酸反应,再酯化,拆分和还原。
此法步骤少,而且产物几乎都为苏式,我国曾采用。
但对硝基苯甲醛用量大,硼氢化钙还存在供应问题。
②与乙醛缩合经对硝基肉桂醇合成氯霉素。
此法使用符合构型要求的反式对硝基肉桂醇为中间体经过溴水加成引入二个官能团,而且产物为苏式。
这条路线的合成步骤不长,而且各步收率不低,是有发展前途的合成方法。
(2) 以苯甲醛为原料硝化时需-20℃低温,限制了此法的应用。
广谱抗菌药氯霉素的合成

氯霉素是一种抑菌类抗生素,由大卫·戈特利布(David Gottlieb)于1947年从南美洲委内瑞拉的土壤内的委内瑞拉链霉菌(Streptomyces venezuelae)成功分离,再于1949年合成并引入临床试验。
氯霉素是世界上首种完全由合成方法大量制造的广谱抗生素,对很多不同种类的微生物均起著作用。
它因价钱低廉的关系,现时仍然盛行于一些低收入国家;但在其他西方国家经已甚少使用,这是由于它的副作用的关系:会引致致命的再生不良性贫血。
现时,氯霉素主要是用在医治细菌性结膜炎的眼药水或药膏上用途氯霉素有着很广泛的活跃性,能有效对抗革兰氏阳性菌,包括大部份的抗药性金黄色葡萄球菌、革兰氏阴性菌及厌氧性生物。
它却不能对抗绿脓杆菌或肠杆菌属的品种。
而对于类鼻疽伯克氏菌,则只有一点效用,但经已由头孢他啶及美罗培南取代。
在西方,氯霉素的使用主要限制在外用上,这是因它有导致再生不良性贫血的风险。
最初氯霉素只用于治疗伤寒,但现时出现能对抗多种药物的伤寒杆菌,显示氯霉素只能抑制某些细菌。
氯霉素亦能用作耐四环素霍乱的第二线治疗。
由于氯霉素对脑脊液高渗透性(远比头孢菌素的高),它是治疗金黄色葡萄球菌引致脑脓疡的第一选择。
它亦可用在治疗由多种生物引致或不明原因的脑脓疡。
氯霉素能有效地抑制三种主要引致脑膜炎的细菌:脑膜炎双球菌、肺炎链球菌及流感嗜血杆菌。
在西方,氯霉素是用于治疗对青霉素或头孢菌素过敏的脑膜炎病人。
而有建议家庭医生须配备有氯霉素的静脉制剂,在一些低收入国家,世界卫生组织亦建议使用氯霉素油剂作为脑膜炎的第一线治疗。
氯霉素的化学名为1R,2R-(-)-1-对硝基苯基-2-二氯乙酰胺基-1,3-丙二醇。
分子中有两个手性碳原子,有四个光学异构体。
其中只有1R,2R-(-)有抗菌活性,即临床使用的氯霉素。
氯霉素为白色或微黄色的针状、长片状结晶或结晶性粉末,味苦。
mp.149~153℃。
本品易溶于甲醇、乙醇、丙酮或丙二醇中,微溶于水。
第七章 氯霉素的合成

在进行异丙醇铝-异丙醇还原时,异丙醇铝的铝原子与羟基上的氧发生配 位结合,同时又与(14-14)分子C-3上的羟基发生脱异丙醇缩合,生成 一种平面式六元环过渡态(14-32),结果生成的产物主要是苏型。
在还原反应中还有副产物“红油”产生,据认为其中 包括一种噁唑环的化合物(14-34)。
在制备异丙醇铝时,加入少量氯化高汞作催化剂,氯 化高汞与铝作用生成铝汞齐以利于迅速开始反应,否 则反应开始缓慢。由于氯化高汞毒性较大,现改用三 氯化铝代替氯化高汞催化反应,取得了同样的效果。 实践证明,采用异丙醇铝与氯代异丙醇铝的混合物比 单独使用异丙醇铝时的收率有较显著的提高。
2. 工艺过程
将对硝基乙苯加入氧化塔中,加入硬脂酸钴及乙酸锰催化剂(内 含载体碳酸钙90%),其量各为对硝基乙苯重量的十万分之五。从塔 底往塔内通进压缩空气,使塔内压力达0.49mPa(5kg/cm2),并调 节尾气压力使达2.9×103Pa(300mm水柱)左右。逐渐升温至 150℃以激发反应,反应开始后,随即发生连锁反应并放热。这时适 当地往反应塔夹层通水使反应温度平稳下降,维持在135℃进行反应。 收集反应生成的水,并根据汽水分离器分出的冷凝水量判断反应进行 的程度。当反应生成热量逐渐减少,生成水的数量和速度降到一定程 度时停止反应,稍冷,将物料放出。 反应物中含对硝基苯乙酮(14-10)、对硝基苯甲酸、未反应的 对硝基乙苯、微量过氧化物以及其它副产物等。在(14-10)未析出 之前,根据反应物的含酸量加入碳酸钠溶液,使对硝基苯甲酸转变为 钠盐。然后充分冷却,使(14-10)尽量析出。过滤,洗去对硝基苯 甲酸钠盐后,干燥,便得(14-10)。
局部溴素过多,则能产生二溴化物(14-26),它不能与六次甲基四 胺成盐。故在下一步成盐反应后二溴化物仍留于溶剂氯苯中。经研究 发现二溴化物(14-26)在溴化氢的催化下能与(14-10)进行反应, 生成2mol的对硝基-α-溴代苯乙酮(14-11)。
氯霉素的合成

氯霉素的合成(总7页)本页仅作为文档页封面,使用时可以删除This document is for reference only-rar21year.March中国矿业大学有机合成与设计A 结课论文论文题目:氯霉素的合成学院:化工学院班级:化工XXX班学号: xxx姓名: XXX2014年6月氯霉素的合成姓名:XXX化工学院化XXX班摘要:氯霉素是一类重要的抑菌抗生素,因其对伤寒病等有疗效,早期得到了大量的应用和发展但是现在医学证明它也存在不小的副作用。
简要的综述了部分经典的合成路线,并分析了各合成路线的优缺点。
关键字:氯霉素、合成路线、立体结构前言氯霉素(chloramphenicol)的化学名D-苏式-(-)-N-[a-(羟基甲基)-β-羟基-对硝基苯乙基]-2,2-二氯乙酰胺。
(1-1)(1-2)本品为白色或微带黄绿色的针状、长片状结晶或结晶性粉末,味苦。
熔点149-153。
本品在甲醇。
乙醇、丙酮或丙二醇中易溶,在水中微溶。
氯霉素(1-1)是广谱抗生素,主要用于伤寒杆菌、痢疾杆菌、脑膜炎球菌、肺炎球菌等感染,对多种厌氧菌感染有效,亦可用于立克次体感染。
本品有引起粒细胞缺乏症及再生障碍性贫血的付能,长期应用可引起二重感染。
新生儿、早产儿用量过大可发生灰色综合症*用约期间必须注意检查血象,如发现轻度粒细胞及血小板减少时,应立即停药。
氯霉素(1-1)发现于1947年,是人类认识的第一个含硝基的天然药物。
1948年用于治疗斑疹伤寒及伤寒。
由于氯霉素的疗效显著,结构较简单,所以发现后就进行了广泛而深人的研究,确定了结构,并根据其结构进行了人工合成及大规模工业生产。
氯霉素(1-1)的化学结构特点是分子中C —1和C —2是两个手性中心,因而它的光学异构体共有4种。
这4种异构体为两对对映异构体,其中一对的构型D —苏型(或称1R ,2R 型,1-2)和L —苏型(或称1S ,2S 型,1-3);另外一对为D-赤型(或称1R ,2S 型,1-4)和L —赤型(或称1S ,2R 型,1-5)。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
实验一 广谱抗菌药氯霉素的合成氯霉素(chloramphenicol )的化学名D-苏式-(-)-N-[a-(羟基甲基)-β-羟基-对硝基苯乙基]-2,2-二氯乙酰胺。
氯霉素(图1,(a ))是广谱抗生素,主要用于伤寒杆菌、痢疾杆菌、脑膜炎球菌、肺炎球菌等感染,对多种厌氧菌感染有效,亦可用于立克次体感染。
氯霉素的化学结构特点是分子中C —1和C —2是两个手性中心,因而它的光学异构体共有4种。
药典收载的本品为D —苏型(1)(图1,(b )),其他三种立体异构体均无疗效。
1.氯霉素合成路线简介氯霉素是人类认识的第一个含硝基的天然药物。
由于氯霉素的疗效显著,结构较简单,所以发现后就进行了广泛而深人的研究,确定了结构,并根据其结构进行了人工合成及大规模工业生产。
我国自五十年代初期从事氯霉素生产以来,基本上采用以乙苯为原料制成对-硝基苯乙-13-酮,然后经溴缩、还原、分拆、二氯乙酞化的“对酮法”工艺路线。
下面选取部分合成路线进行简介。
1.1以具有苯甲基结构的化合物为原料的合成路线1.1.1对硝基苯甲醛与甘氨酸为起始原料的合成路线其合成路线如下: 路线评价:此路线合成步骤少,所需物料品种与设备少;但是,缩合时消耗过量的对硝基苯甲醛,若减少用量,则得到的产物全是不需要的赤型对映体,另外,还需要解决还原剂钙硼氢等原料的来源问题。
图1 (a ) (b )1.1.2对硝基苯甲醛与己醛缩合经对硝基肉桂醇的合成路线对硝基苯甲醛与乙醛进行经羟醛缩合得到对硝基肉桂醛后,采用还原剂将醛还原成醇。
然后从反式对硝基肉桂醇出发经加成、环氧化、L—酒石酸铵拆分等步骤而得氯霉素。
反应路线如下:路线评价:此路线本路线使用符合立体构型要求的反式对硝基肉桂醇为中间体合成步骤不多,各步收率不低是一条有发展前途的合成路线。
1.1.3以苯甲醛为起始原料的合成路线苯甲醛与乙醛反应后再经还原得到肉桂醇,然后经下列过程制得氯霉素。
反应路线如下:路线评价:这条路线的优点是由于最后引入硝基,使得硝化反应中对位体收率高;缺点是,需要在低温下进行,需要制冷设备。
1.2以乙苯为起始原料经对硝基苯乙酮的合成路线1.2.1以乙苯为起始原料经对硝基苯乙酮的合成路线合成路线如下:路线评价:本法的优点是起始原料廉价易得,各步骤反应收率较高,技术要求不高,在巧妙的利用了前手性元素和还原剂的基础上进行手性合成。
缺点是缺点是合成步骤较多,产生大量的中间体和副产物.如无妥善的综合利用途径,必将增加生产负担和巨大的环境污染,对操作者和生产厂家而言,无法回避的就是解决劳动保护和“三废”治理的问题。
1.2.2以乙苯为起始原料对硝基苯乙酮肟的合成路线此合成路线是将对硝基乙苯经亚硝化生成对硝基苯乙酮肟,然后经Neber转位,得到硝基-α-氨基苯乙酮,代替上述反应中的前三步反应。
路线如下:路线评价:本法优点是,硝基乙苯的异构体不需分离,成肟后对位体沉淀析出,而邻位体留在母液中,可省去分离步骤。
缺点是本法工艺过程复杂,原料品种种类较多,而且邻位体的综合利用较困难。
1.3以苯乙烯为起始原料的合成路线1.3.1从苯乙烯出发经α-羟基对硝基苯乙胺的合成路线在氢氧化钠的甲醇溶液中,苯乙烯与氯气反应生成氯代甲醚化物,经硝化反应后,用氨处理得到。
α-羟基对硝基苯乙胺,再经酰化、氧化反应,得到乙酰胺基对硝基苯乙酮,以后步骤与对硝基苯乙酮路线相同。
反应路线如下:路线评价:本法优点是,是原料苯乙烯价廉易得,合成路线较简单且各步收率较高。
若硝化反应采用连续化工艺,则收率高、耗酸少、生产过程安全。
缺点是胺化一步收率不够理想。
1.3.2从苯乙烯出发制成β—卤代苯乙烯经Prins反应的合成路线烯烃与醛(通常是甲醛)在酸的催化下生成1,3—丙二醇及其衍生物的反应,称为Prins 反应,反应的结果不仅在碳链上增加一个碳原于,而且在C—1及C—3上各引入一个羧基。
其路线如下:路线评价:本法优点是,合成步骤较短,从苯乙烯出发经8步反应得到氯霉素,较从乙苯出少了3步反应。
合成路线的前四步中间体均为液体,节省了固体中间分离,干燥,及输送缺点:需用高压反应设备及高真空蒸馏设备。
2.实验室氯霉素合成路线选择综上氯霉素的合成中间体甚多可根据多方面的因素考虑选择合适的路线。
结合实验室的实际情况可以选择以乙苯为起始原料经对硝基苯乙酮的合成路线。
本法的优点是起始原料廉价易得,各步骤反应收率较高,技术要求不高,在巧妙的利用了前手性元素和还原剂的基础上进行手性合成。
合成路线如下:2.1实验方法2.1.1对硝基α-溴代苯乙酮的制备在装有搅拌器、温度计、冷凝管、滴液漏斗的250mL四颈瓶中,加入对硝基苯乙酮10g,氯苯75mL,于25~28℃搅拌使溶解。
从滴液漏斗中滴加溴9.7g。
首先滴加溴2~3滴,反应液即呈棕红色,10min内褪成橙色表示反应开始;继续滴加剩余的溴,约1~1.5h加完,继续搅拌1.5h,反应温度保持在25~28℃。
反应完毕,水泵减压抽去溴化氢约30min,得对硝基α-溴代苯乙酮氯苯溶液,备用。
注释:1.冷凝管口上端装有气体吸收装置,吸收反应中生成的溴化氢。
2.所用仪器应干燥,试剂均需无水。
少量水分将使反应诱导期延长,较多水分甚至导致反应不能进行。
3.若滴加溴后较长时间不反应,可适当提高温度,但不能超过50℃,当反应开始后要立即降低到规定温度。
4.滴加溴的速度不宜太快,滴加速度太快及反应温度过高,不仅使溴积聚易逸出,而且还导致二溴化合物的生成。
5.溴化氢应尽可能除去,以免下步消耗六亚甲基四胺。
2.1.2对硝基α-溴化苯乙酮六亚甲基四胺盐的制备在装有搅拌器、温度计的250mL三颈瓶中,依次加入上步制备好的对硝基α-溴代苯乙酮和氯苯20mL,冷却至15℃以下,在搅拌下加入六亚甲基四胺(乌洛托品)粉末8.5g,温度控制在28℃以下,加毕,加热至35~36℃,保温反应1h,测定终点。
如反应已到终点,继续在35~36℃反应20min,即得对硝基α-溴代苯乙酮六亚甲基四胺盐(简称成盐物),然后冷至16~18℃,备用。
注释:1.此反应需无水条件,所用仪器及原料需经干燥,若有水分带入,易导致产物分解,生成胶状物。
2.反应终点测定:取反应液少许,过滤,取滤液1mL,加入等量4%六亚甲基四胺氯仿溶液,温热片刻,如不呈混浊,表示反应已经完全。
3.对硝基α-溴代苯乙酮六亚甲基四胺盐在空气中及干燥时极易分解,因此制成的复盐应立即进行下步反应,不宜超过12h。
4.复盐成品:mp.118~120℃(分解)。
2.1.3对硝基-α-氨基苯乙酮盐酸盐的制备在上步制备的成盐物氯苯溶液中加入精制食盐3g,浓盐酸17.2mL,冷至6~12℃,搅拌3~5min,使成盐物呈颗粒状,待氯苯溶液澄清分层,分出氯苯。
立即加入乙醇37.7mL,搅拌,加热,0.5h后升温到32~35℃,保温反应5h。
冷至5℃以下,过滤,滤饼转移到烧杯中加水19mL,在32~36℃搅拌30min,再冷至-2℃,过滤,用预冷到2~3℃的6mL乙醇洗涤,抽干,得对硝基-α-氨基苯乙酮盐酸盐(简称水解物),mp.250℃(分解),备用。
注释:1.对硝基-α-溴代苯乙酮与六亚甲基四胺(乌洛托品)反应生成季铵盐,然后在酸性条件下水解成对硝基-α-氨基苯乙酮盐酸盐。
该反应称Delepine反应。
2.加入精盐在于减小对硝基-α-氨基苯乙酮盐酸盐的溶解度。
3.成盐物水解要保持足够的酸度,所以与盐酸的摩尔比应在3以上。
用量不仅导致生成醛等副反应(Sommolet反应),而且对硝基-α-氨基苯乙酮游离碱本身亦不稳定,可发生双分子缩合,然后在空气中氧化成紫红色吡嗪化合物。
此外,为保持水解液有足够酸度,应先加盐酸后加乙醇,以免生成醛等副反应。
4.温度过高也易发生副反应,增加醛等副产物的生成。
2.1.4对硝基-α-乙酰胺基苯乙酮的制备在装有搅拌器、回流冷凝器、温度计和滴液漏斗的250mL四颈瓶中,放入上步制得的水解物及水20mL,搅拌均匀后冷至0~5℃。
在搅拌下加入醋酐9mL。
另取40%的醋酸钠溶液29mL,用滴液漏斗在30min内滴入反应液中,滴加时反应温度不超过15℃。
滴毕,升温到14~15℃,搅拌1h(反应液始终保持在pH3.5~4.5),再补加醋酐1mL,搅拌10min,测定终点。
如反应已完全,立即过滤,滤饼用冰水搅成糊状,过滤,用饱和碳酸氢钠溶液中和至pH7.2~7.5,抽滤,再用冰水洗至中性,抽干,得淡黄色结晶(简称乙酰化物),mp.161~163℃。
注释:1.该反应需在酸性条件下(pH3.5~4.5)进行,因此必须先加醋酐,后加醋酸钠溶液,次序不能颠倒。
中和至碱性,于40~45℃温热30min,不2.反应终点测定:取反应液少许,加入NaHCO3应呈红色。
若反应未达终点,可补加适量的醋酐和醋酸钠继续酰化。
3.乙酰化物遇光易变红色,应避光保存。
2.1.5对硝基-α-乙酰胺基-β-羟基苯丙酮的制备在装有搅拌器、回流冷凝管、温度计的250mL三颈瓶中,投入乙酰化物及乙醇15mL,甲饱和溶液调pH7.2~7.5。
搅拌下缓慢升温,大约40min 醛 4.3mL,搅拌均匀后用少量NaHCO3达到32~35℃,再继续升温至36~37℃,直到反应完全。
迅速冷却至0℃,过滤,用25mL冰水分次洗涤,抽滤,干燥得对硝基-α-乙酰胺基-β-羟基苯丙酮(简称缩合物),mp.166~167℃。
注释:1.本反应碱性催化的pH值不宜太高,pH7.2~7.5较适宜。
pH过低反应不易进行,pH大于7.8时有可能与两分子甲醛形成双缩合物。
甲醛的用量对反应也有一定影响,如甲醛过量太多,亦有利于双缩合物的形成;用量过少,可导致一分子甲醛与两分子乙酰化物缩合。
甲醛用量控制在过量40%左右(摩尔比约为1:1.4)为宜。
2.反应温度过高也有双缩合物生成,甚至导致产物脱水形成烯烃。
3.反应终点测定:用玻棒蘸取少许反应液于载玻片上,加水1滴稀释后置显微镜下观察,如仅有羟甲基化合物的方晶而找不到乙酰化物的针晶,即为反应终点(约需3h)。
2.1.6异丙醇铝的制备在装有搅拌器、回流冷凝管、温度计的三颈瓶中依次投入剪碎的铝片 2.7g,无水异丙醇63mL和无水三氯化铝0.3g。
在油浴上回流加热至铝片全部溶解,冷却到室温,备用。
注释:1.所用仪器、试剂均应干燥无水。
2.回流开始要密切注意反应情况,如反应太剧烈,需撤去油浴,必要时采取适当降温措施。
3.如果无水异丙醇、无水三氯化铝质量好,铝片剪得较细,反应很快进行,约需1-2h,即可完成。
2.1.7DL-苏阿糖型-1-对硝基苯基-2-氨基-1,3-丙二醇的制备在上步制备异丙醇铝的三颈瓶中加入无水三氯化铝 1.35g,加热到44~46℃,搅拌30min。
降温到30℃,加入缩合物10g。