ESC2014-肥厚型心肌病诊断和治疗指南
肥厚型心肌病的猝死风险评估及治疗

肥厚型心肌病的猝死风险评估及治疗肥厚型心肌病(HCM)是一种以心肌肥厚为主要特征的常染色体显性遗传病,其在全球范围内构成了沉重的疾病负担。
心源性猝死是HCM患者的首位死亡原因,也是其最严重的并发症之一。
目前,安装植入式心脏转复除颤起搏器被认为是预防心源性猝死最有效的措施。
近年来,随着医学研究的深入和临床诊疗技术的进步,对HCM的认识日益加深,如何优化该病猝死风险的评估与管理仍是临床实践中的重要课题。
肥厚型心肌病(HCM)是一种常见的遗传性心脏病,主要由编码肌小节蛋白或肌小节相关结构蛋白的基因变异所引起,呈常染色体显性遗传。
临床表现以心肌肥厚为主要特征,其中左心室壁受累最为常见,需排除其他心脏疾病、全身性或代谢性疾病导致的心室壁增厚。
HCM是一种全球性的疾病,文献报道其患病率在0.2%~0.5%左右[1],并常导致多种不良事件的发生,其中心源性猝死(SCD)是其首位死亡原因,也是最严重的并发症之一,构成了沉重的疾病负担。
近年来,随着医学研究的深入和临床诊疗技术的进步,对HCM的认识日益加深,如何优化该病猝死风险的评估与管理仍是临床实践中的重要课题。
一、HCM的猝死风险据研究显示,HCM患者心血管死亡的年发生率为1%~2%,其中SCD、心力衰竭和血栓栓塞是其主要死亡原因[2],而SCD则占据首位,也是其最严重的并发症之一。
部分HCM 患者以SCD为首发表现,常由致命性室性心律失常(如心室颤动)所引起[3-4]。
早期研究表明,HCM是年轻运动员SCD的最常见原因之一,占比约为6%~13%[5-6]。
后续研究也提示,SCD主要影响儿童和年龄<30岁的年轻人,且随着年龄的增长,SCD的风险逐渐降低,在60岁以上的患者中较为少见[3,7]。
此外,研究还发现,HCM患者的SCD风险在不同性别或种族间并无显著差异[8-9]。
二、HCM猝死的危险因素SCD的风险评估是HCM患者临床管理中的一个重要组成部分,几十年来,大量的研究聚焦于确定HCM患者SCD的主要危险因素,并据此进行危险分层,从而确定预防及治疗的策略。
肥厚型心肌病的治疗方法现状与进展

肥厚型心肌病的治疗方法现状与进展乔铅;郝应禄;李燕萍【摘要】肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是常见的常染色体显性遗传性心脏病,其病因复杂,临床表现多样,临床特征是左心室非对称性的肥厚,且这种肥厚不能用其他心脏疾病或全身疾病来解释.随着HCM诊治手段的不断发展、更新,对HCM认识也越来越深入,其治疗方法亦在不断发展.目前治疗上仍主要以防治并发症(尤其是心源性猝死)、减轻患者症状、延缓疾病进展为主,包括一般治疗、药物治疗和非药物治疗.通过阅读相关文献,就HCM的治疗方法作一综述.【期刊名称】《临床荟萃》【年(卷),期】2017(032)012【总页数】5页(P1100-1104)【关键词】心肌病,肥大性;治疗【作者】乔铅;郝应禄;李燕萍【作者单位】昆明医科大学第六附属医院玉溪市人民医院心内科,云南玉溪653100;昆明医科大学第六附属医院玉溪市人民医院心内科,云南玉溪 653100;昆明医科大学第六附属医院玉溪市人民医院心内科,云南玉溪 653100【正文语种】中文【中图分类】R542.2肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一种常见的常染色体显性遗传性心脏病,目前普遍认为其病因约半数以上是由于编码心肌肌节蛋白的基因发生突变。
目前的分子遗传学研究表明,至少有40个基因,超过1 400个基因位点突变被证实与HCM的临床表型相关[1-4]。
HCM以左心室肥厚为特征,尤其是室间隔肥厚,常表现为不对称性肥厚,导致左心室充盈及血液流出受阻,舒张期顺应性下降。
根据左心室流出道压力阶差情况,HCM分为梗阻型、非梗阻型和激发梗阻型。
HCM在成人的患病率为0.02%~0.23%,每年的发病率接近每10万人0.3~0.5[5],我国成年人群发病率约为0.08%[6],然而也有说法认为目前HCM的患病率可能比估计的更常见[7]。
HCM临床表现多种多样,部分患者没有或仅有轻微症状,部分患者出现心悸、胸闷、胸痛、头晕、晕厥等症状,严重者出现室性心律失常甚至心源性猝死(sudden cardiac death, SCD)。
可治性罕见病—肥厚型心肌病

可治性罕见病—肥厚型心肌病一、疾病概述肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一种以心室壁肥厚、增生为特征,常为不对成性肥厚并累计室间隔,左心室血液充盈受阻,舒张期顺应性下降,并且不能够被引起心脏负荷异常的其他疾病(如高血压,瓣膜病变)所解释的原发性心肌病[1]。
肥厚型心肌病也被认为是一种基因突变所致的常染色体显性遗传病。
它是影响儿童和青少年的第二大常见心肌病变类型[2,3],也是青少年及运动员心源性猝死的主要原因。
芬兰,澳大利亚和美国的流行病学资料显示每年肥厚型心肌病的发生率0.24/100000~0.47/100 000[2~4]。
欧洲心脏病协会将肥厚型心肌病分为家族性/基因遗传型和非家族性/非基因遗传型,且多数患者均存在家族遗传史。
肥厚型心肌病病因多样,已证实40%~60%患者存在肌节蛋白基因突变,5%~10%患者存在非肌节蛋白相关的基因突变,少数患者由淀粉样变和内分泌紊乱等非遗传学因素引起[5]。
二、临床特征部分肥厚型心肌病患者没有任何临床表现,而在体检中被发现。
有症状者主要表现为呼吸困难、胸闷、胸痛、心悖、晕厥,甚至猝死。
婴幼儿可表现为喂养困难、生长发育迟缓。
儿童肥厚型心肌病的预后主要取决于其病因与确诊年龄[6]。
继发于先天性代谢缺陷或畸形综合征的儿童主要在出生早期发病,5年生存率分别为42%和74%。
患神经肌肉疾病的儿童通常在较大年龄发病,5年生存率可达98%。
病因未明的儿童,出生后1年内确诊发病的患者和1岁以后确诊患者的5年生存率分别为82%和94%[6]。
对于出生后1年内发病的患儿,充血性心力衰竭为引起死亡最常见的原因[6]。
而猝死则是婴幼儿期后肥厚型心肌病患者的主要死亡原因[6]。
三、诊断通常根据婴幼儿的心脏杂音或充血性心力衰竭的症状(包括呼吸困难,喂养困难,多汗和发育停滞)的评估进行肥厚型心肌病的诊断[7]。
这些症状通常发生于合并左心室(常伴随右心室)流出道梗阻,左心收缩功能呈现高动力性,同时左心室舒张功能开始下降的时候[8]。
中国成人肥厚型心肌病诊断与治疗指南2023

中国成人肥厚型心肌病诊断与治疗指南2023中国成人肥厚型心肌病诊断与治疗指南2023引言:心脏疾病是造成人类死亡的主要原因之一,其中心肌病是一种严重的心脏疾病。
成人肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是最常见的遗传性心肌病之一,它主要特征是心肌肥厚和心室结构的改变,导致心肌收缩和舒张功能障碍。
为了规范和统一对成人肥厚型心肌病的诊断和治疗,中国心脏病学会特制定了《中国成人肥厚型心肌病诊断与治疗指南2023》。
一、诊断标准:1. 心电图(ECG):在ECG上显示左心室肥厚特征波,如增高的QRS波群、ST段改变、T波倒置等。
2. 超声心动图(echocardiography):显示心肌厚度增加,肥厚部位主要集中在室间隔;还可测量心室流出道梗阻度,观察心脏功能状态。
3. 心磁共振成像(MRI):能提供更准确的心脏结构和功能信息,尤其适用于无法通过超声心动图明确诊断的患者。
4. 基因检测:对有家族史或高度怀疑遗传性背景的患者,进行基因检测有助于明确疾病。
二、治疗原则:1. 非药物治疗:- 生活方式干预:控制体重,戒烟限酒,避免剧烈运动,保持规律的有氧运动。
- 心理辅导:帮助患者面对疾病,减轻心理压力。
2. 药物治疗:- β受体阻滞剂:减慢心率,降低心肌耗氧量,减轻心肌肥厚及流出道梗阻。
- 利尿剂:适用于合并有充血性心力衰竭和液体潴留的患者。
- 钙拮抗剂:减少心肌肥厚和心室流出道梗阻,改善心脏舒张功能。
3. 心脏手术:- 流出道梗阻消除术:主要用于有症状且药物治疗无效的患者。
- 心脏移植:对晚期病情进展、合并心力衰竭且无法通过其他手段控制症状者考虑心脏移植治疗。
4. 其他治疗措施:- 植入型心律转复除颤器(ICD):对曾经发生过恶性心律失常的患者进行植入,以预防猝死。
- 爱奇立泵(LVAD):适用于晚期病情进展严重合并心力衰竭的患者,以提供血液循环支持。
三、随访与预后:1. 随访:- 定期进行临床检查、心电图、超声心动图等检查;- 注意家族成员的筛查;- 根据患者病情调整药物治疗;- 提供心理支持和教育患者及家属。
《中国成人肥厚型心肌病诊断与治疗指南2023》解读

HCM的诊断
• 超声心动图或者磁共振检查显示左心室舒张末期任意部 位室壁厚度>15 mm可确诊。
• 若致病基因检测阳性者或者遗传受累的家系成员检查发 现左心室壁厚度>13 mm也可确诊。
HCM的治疗
• 治疗原则 • (1)有症状的梗阻性HCM患者,使用药物治疗或侵 入式治疗方式改善症状。 • (2)有症状的非梗阻性HCM患者,主要针对合并症 进行治疗。
HCM的治疗
• 治疗原则 • (3)HCM患者均应常规开展突发性心脏猝死的风险 评估和危险分层,进行相应预防和治疗。 • (4)无症状HCM患者需要定期进行临床评估。
• 治疗目标 • 缓解临床症状,改善心脏功能,延缓疾病进展,减少 疾病死亡。
HCM的治疗
• 药物治疗 • 在无禁忌证情况下,根据心率、血压情况,从小剂量 开始使用β受体阻滞剂,逐步滴定至最大耐受剂量。 • β受体阻滞剂无效或不耐受的患者,可选用非二氢吡 啶钙拮抗剂(如维拉帕米、地尔硫䓬)。 • 对于使用β受体阻滞剂或非二氢吡啶钙拮抗剂后仍有 明显症状的患者,可联用丙吡胺。
HCM的分型
• 根据血流动力学特点分型可分为: • (3)非梗阻性HCM(静息时或激发后LVOTG峰值 均<30 mmHg) • (4)右心室流出道梗阻(峰值压差≥16 mmHg) 。
HCM的诊断
• 家族史 • 家族成员确诊HCM,一级亲属年龄≤50岁发生突发 性心脏猝死、心力衰竭,或有心脏移植及植入型心律 转复除颤治疗史。
《中ห้องสมุดไป่ตู้成人肥厚型心肌病诊断与治疗 指南2023》解读
HCM的定义
• 肥厚型心肌病(HCM)是一种呈常染色体显性遗传的 原发性心肌病。主要由于编码肌小节相关蛋白的基因致 病性变异引起,临床表现以心肌肥厚为突出特征。
肥厚型心肌病的诊断和防治进展

肥厚型心肌病的诊断和防治进展流行病学和病因肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一种病因不明的左室壁增厚且不伴有心腔扩大为特征的心脏疾病(已排除其他可致心肌肥厚的心脏及系统性原因)。
HCM是最常见的遗传性心脏病,可发生于各个年龄阶段,男女发病率相等,约0.2%~0.5%。
HCM可以继发于30多种不同基因的突变,约60%的青少年与成人患者的病因为编码心脏主要肌节蛋白的基因突变,主要表现为常染色体显性遗传,常呈散发型或交替遗传型。
除此之外,约5%~10%成人患者的病因为其他类型的遗传性疾病,包括染色体异常、代谢和神经肌肉遗传病、遗传综合征、线粒体遗传病;也可以继发于非遗传疾病,如衰老或淀粉样变性。
HCM的病理生理学复杂,涉及舒张功能障碍、心肌缺血和重构、流出道梗阻、心律失常和心力衰竭。
临床表现有很强的异质性,可表现为无症状、心绞痛、呼吸困难、运动耐力下降、晕厥和猝死等。
尽管心脏死亡率的绝对风险每年低于1%,但HCM仍然是青年人及运动员心原性猝死的最常见原因。
由于HCM起病隐匿和进展缓慢,对其进行早期诊断和防治可直接改善患者预后。
近年来,基因检测已用于HCM的早期诊断和危险分层,其与辅助生殖技术的结合还可以指导优生优育。
诊断01诊断标准根据最新公布的2014 ESC指南,HCM的诊断标准在成人中为:任意成像(超声心动图、心脏磁共振成像或计算机断层扫描)检测显示,并非完全因心脏负荷异常引起的左心室心肌某节段或多个节段室壁厚度≥15 mm;在儿童中为左心室室壁厚度≥预测平均值+2×标准差。
对于HCM患者的一级亲属,若心脏成像检测发现无其他已知原因的左心室室壁某节段或多个节段厚度≥13 mm,即可确诊HCM。
02基因检测在诊断方面的重要作用通过基因检测方法确定HCM的致病基因,可以为HCM提供分子诊断依据。
目前,已经发现编码心肌肌节蛋白的20多个基因的1 400多个突变,大量线粒体相关基因和修饰蛋白基因也被发现与HCM 存在关联。
中国成人肥厚型心肌病诊断与治疗指南解读ppt课件

9
10
机制目前仍不明确。
有研究者推测基因突变导致肌纤维收缩功能受损,从而代偿性的出 现心肌肥厚和舒张功能障碍;
也有研究者提出基因突变导致钙循环或钙敏感性受扰,能量代谢受 到影响,从而出现心肌肥厚、纤维化、肌纤维排列紊乱及舒张功能 改变。
HCM是青少年和运动员猝死的主要原因之一。
心脏性猝死(sudden cardiac death,SCD)常见于10~35岁的年轻患者, 心力衰竭(心衰)死亡多发生于中年患者,HCM相关的心房颤动(房颤)导致的 卒中则以老年患者多见。
SCD的危险性随年龄增长而逐渐下降,但不会消失。
在三级医疗中心就诊的HCM患者年死亡率为2%~4%,SCD是最常见的死 因之一。
目前认为,HCM是一种以心肌肥厚为特征的心肌疾病,主要表现为 左心室壁增厚,通常指二维超声心动图测量的室间隔或左心室壁厚 度≥15 mm,或者有明确家族史者厚度≥13 mm,通常不伴有左心室 腔的扩大,需排除负荷增加如高血压、主动脉瓣狭窄和先天性主动 脉瓣下隔膜等引起的左心室壁增厚。
4
典型HCM心肌肥厚
胸骨旁左室长轴切面(图A)显示室间隔明显增厚。应用ECHO测量时需避开右室面肌 小梁。
诊断时还需结合左室短轴切面(图B),左室后壁基底段厚度大多正常。
5
流行病学
6
流行病学
一些来源于特定人群的患病率调查发现HCM并不少见。
美国成年人(23~35岁、51~77岁)HCM患病率为200/10万,中国HCM患 病率为80/10万,粗略估算中国成人HCM患者超过100万。
肥厚型心肌病的诊断与治疗
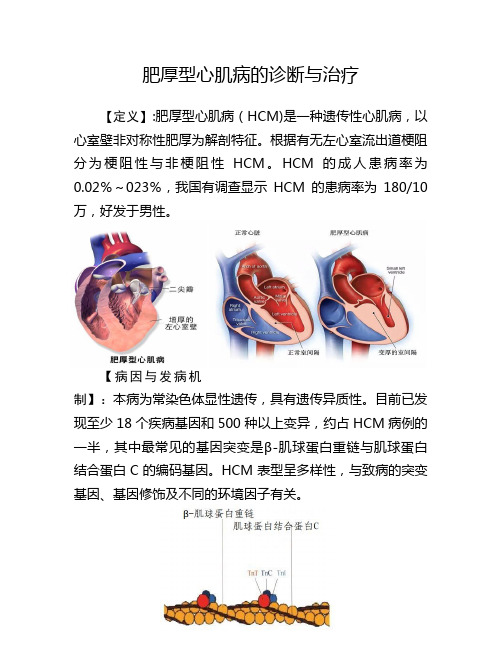
肥厚型心肌病的诊断与治疗【定义】:肥厚型心肌病(HCM)是一种遗传性心肌病,以心室壁非对称性肥厚为解剖特征。
根据有无左心室流出道梗阻分为梗阻性与非梗阻性HCM。
HCM的成人患病率为0.02%~023%,我国有调查显示HCM的患病率为180/10万,好发于男性。
【病因与发病机制】:本病为常染色体显性遗传,具有遗传异质性。
目前已发现至少18个疾病基因和500种以上变异,约占HCM病例的一半,其中最常见的基因突变是β-肌球蛋白重链与肌球蛋白结合蛋白C的编码基因。
HCM表型呈多样性,与致病的突变基因、基因修饰及不同的环境因子有关。
【临床表现】:不同类型病人的临床表现差异较大,半数病人可无症状或体征,尤其是非梗阻型病人。
临床上以梗阻型病人的表现较为突出。
1.症状HCM最常见的症状是劳力性呼吸困难和乏力,1/3的病人有劳力性胸痛,部分病人有晕厥,常于运动时出现,与室性心律失常有关。
该病是青少年和运动员猝死的主要原因。
2.体征主要体征有心脏轻度增大。
梗阻性HCM病人在胸骨左缘第3、4肋间可闻及喷射性收缩期杂音,心尖部也常可闻及收缩期杂音。
增加心肌收缩力或减轻心脏后负荷的措施,如应用正性肌力药物、含服硝酸甘油、Valsalva动作或取站立位均可使杂音增强;相反,使用β受体阳断药、取蹲位等可使杂音减弱。
【实验室及其他检查】:1.X线检查心影正常或左心室增大。
2.心电图主要表现为左心室高电压、ST段压低、倒置T 波和异常Q波。
室内传导阻滞和室性心律失常亦常见。
3.超声心动图是临床最主要的诊断手段。
心室非对称性肥厚而无心室腔增大为其特征。
舒张期室间隔厚度达15mm 或与左心室后壁厚度之比≥1.3。
伴有流出道梗阻的病例可见室间隔流出道部分向左心室突出,左心室顺应性降低致舒张功能障碍。
部分病人心肌肥厚限于心尖部。
4.其他心脏磁共振、心导管检查及心血管造影有助确诊。
心内膜心肌活检可见心肌细胞肥大、排列紊乱、局限性或弥漫性间质纤维化,有助于诊断。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
ESC2014 肥厚型心肌病诊断和治疗指南
肥厚型心肌病(Hypertrophic cardiomyopathy,HCM)是指并非完全因心脏负荷异常引起
的左心室室壁增厚。
虽然 HCM 是一种常见的心血管疾病,但却几乎没有相应的随机对照
临床研究。
所以,本次指南中的大部分推荐都基于观察性队列研究和专家的统一意见,为医生提供所
有年龄段患者临床诊断和治疗概览,此外,由于大部分患者都有遗传病因参与,本次指南
同时还纳入了对家庭成员进行诊断的内容,并对生殖和避孕做出了特别建议。
该定义对儿童与成人均可适用,并不需要对病因或心肌病理进行先验推测。
虽然这个定义
会扩大指南的范围,并使一些推荐复杂化,但与每日临床实践紧密相关,能更好地改善诊
断的准确性和治疗。
一、病因
高达 60% 的青少年与成人 HCM 患者的病因是心脏肌球蛋白基因突变引起的常染色体显性
遗传。
5-10% 的成人患者病因为其他遗传疾病,包括代谢和神经肌肉的遗传病、染色体异
常和遗传综合征。
还有一些患者的病因是类似遗传疾病的非遗传疾病,如老年淀粉样变性。
二、诊断标准
1. 成人
成人中HCM 定义为:任意成像手段(超声心动图、心脏核磁共振成像或计算机断层扫描)检测显示,并非完全因心脏负荷异常引起的左室心肌某节段或多个节段室壁厚度≥ 15 mm。
遗传或非遗传疾病可能表现出来的室壁增厚程度稍弱(13-14 mm),对于这部分患者,需要评估其他特征以诊断是否为 HCM,评估内容包括家族病史、非心脏性症状和迹象、心
电图异常、实验室检查和多模式心脏成像。
2. 儿童
与成人中一样,诊断 HCM 需要保证 LV 室壁厚度≥ 预测平均值+ 2 SD(即 Z 值>2,Z 值
定义为所测数值偏离平均值的 SD 数量)。
3. 亲属
对于 HCM 患者的一级亲属,若心脏成像(超声心动图、心脏核磁共振或 CT)检测发现无其他已知原因的 LV 室壁某节段或多个节段厚度≥ 13 mm,即可确诊 HCM。
在遗传性 HCM 家族中,未出现形态学异常的突变携带者可能会出现心电图异常,这种异
常的特异性较差,但在有遗传性 HCM 的家族成员身上,可视为 HCM 疾病的早期或温和
表现,其他多种症状也可以提高对这部分人群诊断的准确性。
总而言之,对于遗传性HCM 的家族成员,任何异常(如心肌多普勒成像和应变成像异常、不完全二尖瓣收缩期前移或延长和乳头肌异常)尤其是心电图异常都会大大增加该成员诊
断出 HCM 的可能性。
三、诊断
1. 静息和动态心电图检查建议
2. 超声心动图检查建议
(1)经胸超声心动图检查建议
(2)经食管超声心动图检查建议
3. 心血管核磁共振成像检查建议
4. 核成像和计算机断层成像
5. 心内膜心肌活检
6. 遗传学检测和家族筛选
(1)遗传咨询建议
(2)对先证者进行遗传检测的建议
(3)成年亲属遗传和临床检查推荐
(4)对儿童进行遗传检测和临床检查的建议
(5)对无表型突变携带者的随访建议
四、症状评估
1. 胸痛
冠脉血管造影术检查建议
2. 心衰
(1)侵入性血流动力学研究建议
(2)心肺运动试验建议
3. 晕厥
4. 心悸
电生理检查建议
五、症状和并发症治疗
1. 左心室流出道阻塞(LVOTO)(1)LVOTO 治疗建议:一般措施
(2)药物治疗建议
(3)室间隔消融治疗建议
(4)起搏治疗建议
2. 左心室流出道未阻塞患者的症状治疗
(1)LV 射血分数正常(≥ 50%)的心衰患者治疗建议
(2)LV 射血分数降低的心衰患者治疗建议
(3)心脏再同步化治疗建议
(4)心脏移植建议
(5)左心室辅助装置建议
(6)无左心室流出道阻塞、用力时胸痛患者的治疗建议。