丁三烯、环丙烷与双键的[3+2]环加成反应方法学研究
功能化分子筛催化氮杂环丙烷与异硫氰酸酯[3+2]环加成反应的研究
![功能化分子筛催化氮杂环丙烷与异硫氰酸酯[3+2]环加成反应的研究](https://img.taocdn.com/s3/m/20ee17c1185f312b3169a45177232f60dccce75b.png)
功能化分子筛催化氮杂环丙烷与异硫氰酸酯[3+2]环加成反应的研究任鸿;盛丽丽【摘要】主要研究了固体酸催化剂对氮杂环丙烷与异硫氰酸酯间的[3+2]环加成反应的催化效果,并对反应展开了一系列底物拓展.实验结果表明,在该催化体系下,反应都有不错的收率.并且通过催化剂重复性实验,也可以发现,经过多次重复利用的固体酸也可以起到有效的催化作用.【期刊名称】《浙江化工》【年(卷),期】2019(050)007【总页数】4页(P16-19)【关键词】氮杂环丙烷;异硫氰酸酯;功能化分子筛;环加成反应【作者】任鸿;盛丽丽【作者单位】浙江省疾病预防控制中心, 浙江杭州 310051;浙江省安全生产科学研究院, 浙江杭州 310012【正文语种】中文0 引言氮杂环丙烷类化合物由于其具有较高的环张力,因而在有机合成中,基于氮杂环丙烷开环的反应应用非常广泛[1]。
氮杂环丙烷与聚集双键化合物如异氰酸酯和硫异氰酸酯的[3+2]环加成反应,可以得到一系列功能化的五元杂环,对于生物化学及药物化学都有非常广泛的应用。
经过多年研究,一系列催化体系被开发并应用于该反应上,比如 Pd[2]、Ni[3]、HBF4[4]、PBu3[5]、NaI[6]和 Ph4Sb-Br[7]等。
然而这些反应体系多数需要在惰性气体的保护下进行,同时上述反应多为均相催化体系,反应后处理较为繁琐。
分子筛因其具有独特的孔结构,以及不挥发性、高稳定性、与产物分离简便等优点,被广泛应用于酸催化反应中。
因此,我们希望将其特性应用到氮杂环丙烷的环加成反应中,以担载杂多酸的固体酸做催化剂,进一步提高其催化活性,并能多次重复利用,如图1所示。
图1 SBA-15-PW催化氮杂环丙烷与硫异氰酸酯的反应Fig.1 Reaction of aziridines with isothiocyanates catalysted by SBA-15-PW1 氮杂环丙烷的合成一般而言,氮杂环丙烷的合成途径主要有以下途径,首先,氮宾对烯烃的加成;其次,卡宾或叶立德试剂对亚胺的加成;还有就是1,2-氨基醇或1,2-卤代氨基等邻氨基化合物的环化反应。
金鸡纳碱类化合物催化的不对称[3+2]成环反应研究的开题报告
![金鸡纳碱类化合物催化的不对称[3+2]成环反应研究的开题报告](https://img.taocdn.com/s3/m/b458eff96037ee06eff9aef8941ea76e58fa4ac7.png)
金鸡纳碱类化合物催化的不对称[3+2]成环反应研究
的开题报告
一、研究背景与意义
不对称的合成方法在新药物合成、天然产物合成以及功能性材料合成等领域中都具有重要意义。
其中,金鸡纳碱类化合物催化的不对称[3+2]成环反应成为了一种重要的不对称合成手段。
该反应是利用稠环单碳杂环化合物与烯丙基酮类化合物反应,生成具有高立体选择性的四元杂环化合物。
本研究旨在进一步研究金鸡纳碱类化合物催化的不对称[3+2]成环反应,对其反应机理以及反应条件进行深入研究,为其在不对称合成领域中的应用提供更多的理论支持和实验基础。
二、研究内容与方法
本研究将探索不同结构的金鸡纳碱类化合物对于[3+2]成环反应的催化效果,研究催化剂种类和配体结构对于立体选择性的影响。
同时,将探究不同反应条件对于催化剂活性和立体选择性的影响,比如反应底物的化学结构、反应溶剂、反应温度和反应时间等。
研究方法包括催化反应实验、NMR谱图分析、IR光谱分析、质谱分析等。
三、研究预期结果
本研究预计能够研究得到金鸡纳碱类化合物催化的不对称[3+2]成环反应的反应机理以及催化机理,并且能够发现催化剂种类和配体结构对于立体选择性的影响。
同时,对于反应条件的优化,预计能够获得反应条件下的最佳催化效果。
该研究可以为金鸡纳碱类化合物催化的不对称合成方法提供更多的理论和实验支持。
金属试剂参与的环丙烯开环反应研究

金属试剂参与的环丙烯开环反应研究环丙烯开环反应是一种重要的催化反应,其中金属试剂参与是其关键步骤。
近年来,这类反应已被广泛用于合成医药、农药、色素和其他有用化合物。
本文将对金属试剂参与的环丙烯开环反应研究进行详细描述,并综合分析金属试剂参与的环丙烯开环反应的结果。
环丙烯开环反应是一种双加成反应,它的反应方程式为:环丙烯 +成剂丙酮 +一种产物金属试剂在这类反应中起着重要作用,改变其反应速率,改变其反应条件,以及影响其产物组成等。
有很多金属试剂,如钴,镍,铜,锰和铁等,可以用来催化环丙烯开环反应。
钴催化环丙烯开环反应是一种重要的反应技术。
研究发现,正交钴试剂(Co-DPPA)通过钴催化反应,可以有效地将环丙烯开环为环丙酮和另一种产物。
实验发现,使用正交钴试剂,反应温度可以从常温降低到-20°C,反应时间可以从数小时减少到数分钟,反应产率可以达到90%以上。
此外,钴催化环丙烯开环反应可以实现高度选择性,可以有效地防止不需要的端点反应。
除钴催化以外,还有许多其他金属试剂可以用来催化环丙烯开环反应,其中镍催化是最重要的一种。
研究发现,使用镍催化剂可以将环丙烯开环为环丙酮和2-氨基丙酸酯的类硫醚的混合物,反应产率可以达到80%。
这种反应可以在一般常温条件下进行,但不需要使用过量的镍催化剂,反应时间相对较长,只有2小时。
除了镍和钴催化以外,铜,锰和铁等也可以用来催化环丙烯开环反应,但是它们的效果没有钴和镍那么明显。
铜催化的反应产率可以达到50%,反应时间也比钴镍催化长一些,只需要3小时。
锰催化的反应产率也只有25%,反应时间也比钴镍催化稍长,为4小时。
铁催化的反应产率最低,只有20%,反应时间也比钴镍催化要长,只有5小时。
从上面可以看出,金属试剂参与的环丙烯开环反应有许多种,其中钴和镍催化的反应产率最高,反应时间最短,可以有效节省时间和能量,是最重要的反应技术之一。
但是,这些金属试剂的反应结果还要受到反应条件的影响,比如反应温度、反应溶剂、反应时间等。
环加成反应机理计算课题组

环加成反应机理计算课题组
环加成反应是有机化学中的一类重要反应,通常是指通过环状化合物的加成反应,使分子中的某个环被打开,然后重新形成一个新的环。
这种反应机理的计算对于理解反应的具体步骤和确定反应的产物具有重要意义。
环加成反应机理计算课题组是一个研究团队或课题组,致力于使用计算化学的方法和模拟技术来研究环加成反应的机理。
该课题组可能包括有机化学家、计算化学家和理论化学家等不同领域的专家。
这个课题组可能会使用各种计算方法,如量子力学计算、分子模拟和密度泛函理论等,来探索环加成反应的反应路径、中间体和过渡态结构,并通过计算得出反应的热力学和动力学参数。
通过研究环加成反应的机理,课题组可以为有机合成化学提供新的反应途径和方法,并为设计新的催化剂和化学反应体系提供理论指导。
此外,他们的研究还能帮助解释实验中观察到的反应现象,并为合成新药物和功能材料等领域的研究提供理论支持。
总之,环加成反应机理计算课题组的研究对于理解有机反应机理、推动有机合成化学的发展以及解决实际问题具有重要意义。
环加成反应和机理精品PPT课件
讲师:XXXXXX XX年XX月XX日
FMO理论认为,在双分子热反应中,起决定作用的是前线 分子,即一分子出LUMO,另一分子出 HOMO。两个起作用的 轨道必须具有相同的对称性且能量相近才能重叠。
一般环加成为同面环加成
同面环加成
(suprafacial cycloaddition)
异面环加成
(antarafacial cycloaddition)
SOMO'= ψ1 S
S SOMO'= ψ1
对称性匹配 可以成环
对称禁阻的[2 π +2 ห้องสมุดไป่ตู้]热环化加成反应
前线轨道 (FMO)理论认为,在双分子光反应中,两 组分均为具有两个成单电子的激发态分子,单电子占据 的MO又称为SOMO,。故光照下的环加成方式为: 两组分能量较高的两个SOMO组合形成一个σ单键; 两组分能量较低的两个SOMO组合形成另一个σ单键。 两组分相互组合的SOMO必须具有相同的对称性且能量相 近才能重叠。若对称性不同则不能发生环加成反应
环化反应: 加成反应:
环加成反应
+
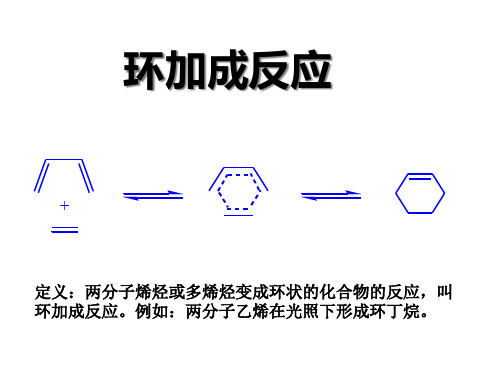
迁移反应: 定义:两分子烯烃或多烯烃变成环状的化合物的反应,叫 环加成反应。例如:两分子乙烯在光照下形成环丁烷。
环加成反应类型
[2+1]环加成: CH2 CH2
[2+2]环加成: CH2 CH2
R2C:
RR
CH2 CH2
[4+1]环加成: CH2 CH CH CH2 SO2
对称允许的[4π+2π]热环化加成反应
Ψ2(A) Ψ2(A)
Ψ3(S) Ψ1(S)
烯烃与联烯的不对称[2+2]环加成反应
![烯烃与联烯的不对称[2+2]环加成反应](https://img.taocdn.com/s3/m/b5442790b8f3f90f76c66137ee06eff9aef849d8.png)
烯烃与联烯的不对称[2+2]环加成反应一、概述1.1 烯烃与联烯的不对称[2+2]环加成反应概述烯烃与联烯的不对称[2+2]环加成反应是有机化学领域中的重要研究内容之一。
该反应以其高效、高立体选择性等特点备受关注,并且在天然产物和药物合成中具有广泛的应用前景。
本文将对烯烃与联烯的不对称[2+2]环加成反应进行系统的介绍和总结,力求全面、客观地展现该领域的研究进展。
1.2 研究目的和意义本文旨在系统介绍烯烃与联烯的不对称[2+2]环加成反应的研究现状,包括其反应机理、影响因素、反应条件控制等方面,并对相关研究的进展和应用进行分析和总结。
通过本文的阐述,可以更好地理解该反应的原理和特点,为进一步的研究和应用提供有益的参考。
二、烯烃与联烯的不对称[2+2]环加成反应的研究现状2.1 反应机理烯烃与联烯的不对称[2+2]环加成反应是通过自由基、金属催化等多种机理进行的,不同的反应条件会导致不同的机理途径。
研究者们通过实验和理论计算等手段,对不同条件下的反应机理进行了深入探讨,为该反应的合理设计和优化提供了重要的理论基础。
2.2 影响因素在进行烯烃与联烯的不对称[2+2]环加成反应时,反应条件的选择和控制对反应结果具有重要的影响。
催化剂的选择、温度和溶剂等因素都会对反应产物的立体化学和产率产生显著影响。
研究者们对这些影响因素进行了系统的分析和研究,为优化反应条件提供了重要的参考依据。
2.3 应用前景由于烯烃与联烯的不对称[2+2]环加成反应具有高效、高立体选择性等特点,因此在天然产物和药物合成中有着广泛的应用前景。
研究者们通过不断的改进和优化,已经取得了许多重要的实验结果,为该反应的工业化应用奠定了良好的基础。
三、研究展望3.1 反应条件的优化随着研究的不断深入,烯烃与联烯的不对称[2+2]环加成反应的反应条件得到了不断的优化。
未来的研究将进一步探索新的催化剂、新的溶剂体系和新的反应条件,以实现该反应的更高效、更高立体选择性的进行。
丁三烯、环丙烷与双键的环加成反应方法学研究

丁三烯、环丙烷与双键的[3+2]环加成反应方法学研究本文主要研究了丁三烯与双键的[3+2]环加成反应、DA(donor-acceptor)-环丙烷与羰基的交叉[3+2]环加成串联反应、以及DA-环丙烷与异氰酸酯的活性碳氮键的平行[3+2]环加成串联反应。
同时也对麦角类生物碱的不对称全合成进行了探索,全文共分为五章:第一章综述了[3+2]环加成反应的研究进展,对1,3-偶极[3+2]环加成反应,过渡金属催化的[3+2]环加成反应,丙二烯的[3+2]环加成反应,以及DA-环丙烷的开环[3+2]环加成反应进行了全面总结。
第二章在总结累积烯烃与芳香醛的反应进展以及丁三烯参与的反应类型基础上,发展了PBu3催化的丁三烯分别与芳香醛或α,β不饱和羰基化合物之间的两类区域选择性[3+2]环加成反应,构筑了一系列多取代2,5-二氢呋喃衍生物或含有季碳中心的环戊烯类衍生物。
两类反应表现出不同的区域选择性:前者是由丁三烯的α位首先进攻芳香醛的羰基进而关环,生成多取代2,5-二氢呋喃衍生物,而后者是通过丁三烯的γ位首先进攻α,β-不饱和羰基化合物的双键进而关环,生成含有季碳中心的环戊烯类衍生物。
机理研究证明不同的区域选择性主要是受底物分子之间空间位阻的控制。
第三章基于对构筑含有桥氧复杂结构方法的总结,发展了一类全新的Lewis acid 催化的环氧重排/DA-环丙烷与醛酮的分子内交叉[3+2]环加成串联反应,该反应在温和条件下可一步构筑一系列含有复杂并环或螺环结构的桥氧-[n.2.1]骨架。
其中与DA-环丙烷发生交叉[3+2]环加成反应所需的羰基是通过环氧重排反应原位生成,用普通方法不易制备。
该串联反应普适性强,对脂肪族和芳香族环氧底物都适应。
通过机理研究证明反应表现出单一的非对映选择性是受Lewis acid与底物的络合作用控制。
第四章在总结构筑吡咯[1,2-a]吲哚骨架的基础上,成功发展了一类Curtius重排/DA-环丙烷与异氰酸酯的活性碳氮键的分子内平行[3+2]环加成串联反应,高效构筑了一系列含有吡咯[1,2-a]吲哚骨架的化合物。
环加成反应

化学术语
01 反应过程
03 反应原理
目录
02 反应实例
环加成反应,cycloaddition reaction
两个共轭体系结合成环状分子的一种双分子反应。通过环加成反应,两个共轭体系分子的端基碳原子彼此头 尾相接,形成两个σ键,使这两个分子结合成一个较大的环状分子,例如丁二烯与乙烯(或它们的衍生物)的加 成反应。
在有机化学中我们已经知道,两个分子中的轨道相互作用,必然产生两个新的分子轨道,一个轨的能量降 低△E,另一个轨道的能量升高△E,由于反键效应, △E略大于△E。当两个HOMO轨道相互作用时,结果使总的 分子轨道的能量增加,体系更加不稳定,因而HOMO轨道件无相互作用,不能成键。
当HOMO轨道与LUMO轨道相互作用时,形成两个新的轨道,一个能量降低,较HOMO轨道低,另一个轨道能量升 高,较LUMO轨道高,生电子优先排入能量较低的轨道,使整体能量降低,体系趋于稳定,因而可以成键。
2、能量相近规则。相互作用的HOMO和LUMO轨道,能量必须接近,能量越接近,反应越容易进行。两轨道能 量越接近,形成成键轨道能量越低,成键后能量降低的更多,体系更稳定。
3、轨道最大重叠规则。在双分子环加成反应过程中,电子云密度大的原子倾向于与电子云密度大的原子相连, 以这种方式成键后新化学键的键能更高,体系更稳定。
前线轨道
处理原则
分子周围的电子云,根据能量的不同,可以分为不同的能级轨道,根据能量最低原理,电子优先排入能量低 的轨道。前线轨道理论中,将占有电子的能级最高的轨道称之为最高占有轨道,用HOMO表示;未占有电子的能量 最低的轨道称之为最低占有轨道,用LUMO表示。有的共轭轨道中含有奇数个电子,它的最高已占有轨道只有一个 电子,这种单电子占有的轨道称之为单占轨道,用SOMO表示。在分子中,HOMO轨道对于电子的束缚最为薄弱, LUMO轨道对电子的吸引力最强,因而前线轨道认为,分子加发生化学反应,本质上就是HOMO轨道与LUMO轨道的相 互作用,形成新的化学键的过程。特别的,SOMO在前线轨道理论中即可作为HOMO处理,也可作为LUMO处理。我们 将HOMO轨道和LUMO轨道统称为前线轨道,用FOMO表示,前线轨道上的电子称为前线电子。所以,在分子间化学反 应过程中,最先作用的轨道是前线轨道,起关键作用的电子为前线电子。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
丁三烯、环丙烷与双键的[3+2]环加成反应方法学研究
本文主要研究了丁三烯与双键的[3+2]环加成反应、DA(donor-acceptor)-环丙烷与羰基的交叉[3+2]环加成串联反应、以及DA-环丙烷与异氰酸酯的活性碳氮键的平行[3+2]环加成串联反应。
同时也对麦角类生物碱的不对称全合成进行了探索,全文共分为五章:第一章综述了[3+2]环加成反应的研究进展,对1,3-偶极[3+2]环加成反应,过渡金属催化的[3+2]环加成反应,丙二烯的[3+2]环加成反应,以及DA-环丙烷的开环[3+2]环加成反应进行了全面总结。
第二章在总结累积烯烃与芳香醛的反应进展以及丁三烯参与的反应类型基础上,发展了 PBu3催化的丁三烯分别与芳香醛或α,β不饱和羰基化合物之间
的两类区域选择性[3+2]环加成反应,构筑了一系列多取代2,5-二氢呋喃衍生物或含有季碳中心的环戊烯类衍生物。
两类反应表现出不同的区域选择性:前者是由丁三烯的α位首先进攻芳香醛的羰基进而关环,生成多取代2,5-二氢呋喃衍
生物,而后者是通过丁三烯的γ位首先进攻α,β-不饱和羰基化合物的双键进而关环,生成含有季碳中心的环戊烯类衍生物。
机理研究证明不同的区域选择性主要是受底物分子之间空间位阻的控制。
第三章基于对构筑含有桥氧复杂结构方法的总结,发展了一类全新的Lewis acid
催化的环氧重排/DA-环丙烷与醛酮的分子内交叉[3+2]环加成串联反应,该反应在温和条件下可一步构筑一系列含有复杂并环或螺环结构的桥氧-[n.2.1]骨架。
其中与DA-环丙烷发生交叉[3+2]环加成反应所需的羰基是通过环氧重排反应原位生成,用普通方法不易制备。
该串联反应普适性强,对脂肪族和芳香族环氧底物都适应。
通过机理研究证明反应表现出单一的非对映选择性是受Lewis acid与底物
的络合作用控制。
第四章在总结构筑吡咯[1,2-a]吲哚骨架的基础上,成功发展了一类Curtius重排/DA-环丙烷与异氰酸酯的活性碳氮键的分子内平行[3+2]环加成串联反应,高效构筑了一系列含有吡咯[1,2-a]吲哚骨架的化合物。
其中与DA-环丙烷发生平行[3+2]环加成反应所需的异氰酸酯是通过
Curtius重排反应原位生成,用普通方法不易制备。
DA-环丙烷与异氰酸酯的反应表现出单一的区域选择性。
第五章制备了手性炔丙醇S-(+)-5-3,并以它为关键中间体探索了麦角碱(+)-cycloclavine的不对称全合成,实现了手性传递的aza-Cope-Mannich反应,同时设计了串联反应策略探索(+)-cycloclavine的全合成。
针对麦角碱fumigaclavine A的全合成制备了关键中间体5-47,为完成其全合成奠定了基础。
全文对[3+2]环加成反应的合成方法学进行了拓展,完成了不同类型环状化合物的构筑,为构建复杂结构天然产物的骨架提供了高效的合成策略。
也对麦角碱类天然产物全合成进行了有益的探索。