盐酸左旋咪唑片的含量测定
盐酸左旋咪唑中对甲苯磺酰氯杂质测定方法的建立
2 . 4 线性范 围
1 . 3药品 :盐酸左旋 咪唑 L V 1 2 5 0 4 。 2 方法 和结 果 2 . 1色谱 条件
色 谱 仪 :岛津L C 一 2 0 A T 或 相 当 的液 相 色谱 系 统 。色 谱 柱 :C 。
国眶|雷—盈同
2 0 1 4 年7 月第 1 2卷 第 1 9期
・
实验研 究 ・ l 0 7
盐 酸左旋 咪唑 中对 甲苯磺酰氯杂质 测定 方法的建 立
邢 亚 非 钱 小 燕
( 南京 白敬宇制药有 限责任公 司,江苏 南京 2 1 0 0 3 8 )
【 摘 要 】 目的 建立 盐 酸左 旋咪 唑 中对 甲苯磺 酰 氯杂质 测 定方 法并 对方 法进 行验 证 。方法 色谱柱 c ( 5 m× 4 . 61 T I r f l × 2 5 0 m m) , 流 动概
呈 良好 线性 关 系,相 关 系数 R 2≥ 0 . 9 9 。结 论 该方 法 专属性 强,重 复性好 ,可 用于 盐酸左 旋咪 唑 中对 甲苯磺 酰 氟 的定量 测定 。 【 关 健 词】 盐 酸左 旋咪 唑 ;对 甲苯磺 酰 氯 ;高效液 相 色谱 法
中图分 类号 :R 9 2 7 . 2
试液浓度 1 mg / m L ) 2 . 3 . 2 检测 限与定 量 限 :取对 照品溶 液逐级稀 释 ,定量 限浓度 为对 甲
磷 酸 试剂级 甲醇 色谱纯 盐酸左旋咪唑 供 试品
来源 百灵威 科技有限公司 南京 白敬宇制药有限责任公司 南京化学 试剂有限责任公司 国药集 团化学 试剂有 限公司
美 国T E D I A试剂有限公司 南京 白敬宇制药有限责任公 司
复方盐酸左旋咪唑注射液含量测定方法的建立

5 0 m L容量瓶 中用 水稀 释至 刻度线 ,摇匀 即得 。
2 . 2 . 2 供试 品溶液 的制备 取本 品 2Байду номын сангаасm L( 约相 当
于 盐 酸左 旋 咪 唑 5 0 mg ) ,置 5 0 mL容 量 瓶 中 ,用
表 1 复 方 盐酸 左 旋 咪 唑 注射 液 中黄 芪 多 糖 的 平均 含 量
中 ,用 水稀 释 至刻 度 ,摇 匀 ,再 吸取 1 0 mL置于
乙胺调节 p H值至 4 . 0 - 4 - 0 . 0 5 )一乙腈 ( 8 O: 2 0 ) ;
流速为 1 . 0 mL / mi n ;进 样 量 为 2 0 L 。在 上 述 色
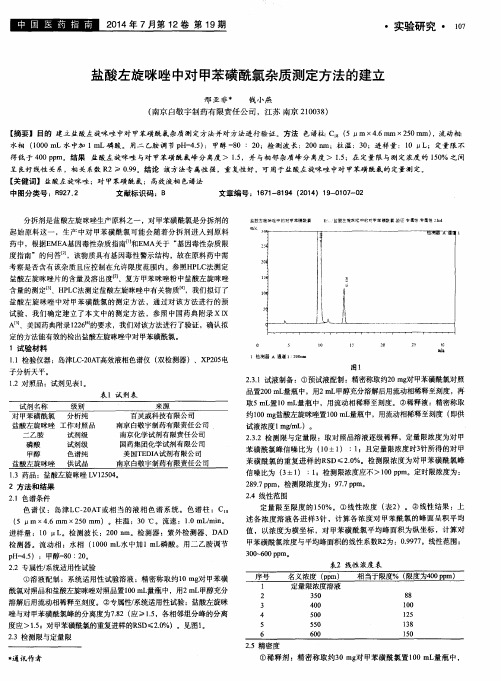
谱条件下 ,盐酸左旋 咪唑峰与杂峰的分离度符 合要求 ,保 留时间为 2 . 7 m i n 左右 ,保 留时间适 中 ,色谱 图见图 1 ;另外 ,波长扫描 图谱 见 图
究
2 . 1 黄芪 多糖含 量测 定
2 . 1 . 1 对 照 品溶 液 的制 备 取 在 1 0 5  ̄ C干 燥 至 恒 重 的无 水 葡 萄 糖 O . 1 g ,精 密 称 定 ,至 1 0 0 mL容 量 瓶 中 ,加 水 溶 解并 稀 释 至 刻度 线 ,摇 匀 ;精 密量 取 l O m L ,置 1 0 0 mL容 量瓶 中 ,加 水 至刻度
线 ,摇匀 ,即得 。
其吞噬 、趋化功能 ,诱导机体产生各种 细胞 因 子 ;提 高补体 活性 ,增强机 体 巨噬细胞功 能 , 提高抗 体产生水 平 ,可用 于动物 的抗 菌消 炎 、 抗病毒 、抑制肿瘤和促进生长等方面_ 3 _ 。二者合
用 ,杀 虫 的 同时 增 强 机 体 免 疫 功 能 、提 高 机 体 抵抗 力 ,效 果 明显 。
(原料模版)STP-ZL-351-01 盐酸左旋咪唑内控质量标准

盐酸左旋咪唑内控质量标准文件类别STP 文件编码版本号ⅡSTP-ZL-351-01 起草/修订人日期审核人日期批准人日期生效日期颁发部门行政部分发部门:质量部一、目的:本程序规定了盐酸左旋咪唑的质量标准。
二、适用范围:适用于盐酸左旋咪唑的检验、放行。
三、职责:QA人员:负责按要求起草;质量部经理:负责本STP审核;总经理:批准本STP的执行。
四、正文:1 【标准依据】《中国兽药典》2010版一部P187。
2 【内控质量标准】盐酸左旋咪唑Yansuan ZuoxuanmizuoLevamisole HydrochlorideC11H12N2S〃HCl 240.76本品为(S)-(-)-6-苯基-2,3,5,6-四氢咪唑并[2,1-b]噻唑盐酸盐。
按干燥品计算,含C11H12N2S〃HCl不得少于98.5%。
【性状】本品为白色或类白色的针状结晶或结晶性粉末;无臭,味苦。
本品在水中极易溶解,在乙醇中易溶,在三氯甲烷中微溶,在丙酮中极微溶解。
熔点本品的熔点(附录51页)为225~230℃。
比旋度取本品适量,精密称定,加水溶解并定量稀释制成每1ml中约含50mg的溶液。
依法测定(附录53页),比旋度为不低于-121. 5°。
【鉴别】(1)取本品约60mg,加水20ml溶解后,加氢氧化钠试液2ml,煮沸10分钟,放冷,加亚硝基铁氰化钠试液数滴,即显红色;放臵后,色渐变浅。
(2)本品的红外光吸收图谱应与对照的图谱一致。
(3)本品水溶液显氯化物的鉴别反应(附录25页)。
【检查】溶液的澄清度取本品2.0g,加新沸并冷至20~25℃的水50ml,溶解后,溶液应澄清;如显浑浊,与2号浊度标准液(附录95页)比较,不得更浓。
酸度取溶液的澄清度项下的溶液,依法测定(附录56页),pH值应为3.5~5.0。
吸光度取本品,加盐酸甲醇滴定液(0.2mol/L)制成每1ml中含1 mg的溶液,照紫外-可见分光光度法(附录26页),在310nm的波长处测定,吸光度不得过0.20。
HPLC法测定盐酸左旋咪唑糖浆的含量

HPLC法测定盐酸左旋咪唑糖浆的含量
李洁;林慧菁
【期刊名称】《中国现代药物应用》
【年(卷),期】2009(003)019
【摘要】目的建立高效液相色谱法测定盐酸左旋咪唑糖浆含量的方法.方法色谱柱为Agilent-C18(250 mm×4.6 mm,5 μm);流动相为0.025 mol/L枸橼酸(用三乙胺调节pH值为4.5)-乙腈(85:15);检测波长为277 nm;流速为1.0 ml/min.结果盐酸左旋咪唑在5~100 μg/ml范围内有良好的线性关系(r=0.9998),平均回收率(n=9)为100.9%(RSD=1.4%).结论本法简单、快速、准确,可用于盐酸左旋咪唑糖浆的定量分析.
【总页数】2页(P22-23)
【作者】李洁;林慧菁
【作者单位】523109,广东省东莞市药品检验所;523109,广东省东莞市药品检验所【正文语种】中文
【相关文献】
1.HPLC法测定咳特灵糖浆中马来酸氯苯那敏的含量及含量均匀度 [J], 江溱
2.HPLC法测定复方甲苯咪唑片中甲苯咪唑和盐酸左旋咪唑的含量 [J], 郭江红;赵亚萍
3.提取旋光法测定糖浆中盐酸左旋咪唑含量 [J], 杨燕;王屹澄
4.HPLC法测定盐酸左旋咪唑片的含量研究 [J], 周芷锦;林仙军;王彬;陈晓林
5.HPLC法测定复方氨酚美沙糖浆中对氨基酚含量 [J], 蔡锦雄;江坤;陈颜清;刘敏;李玉兰
因版权原因,仅展示原文概要,查看原文内容请购买。
用GC法测定盐酸左旋咪唑在驱虫药中的含量

用GC法测定盐酸左旋咪唑在驱虫药中的含量摘要:目的:测定驱虫药复方鹧鸪菜散中盐酸左旋咪唑的含量,以进一步提高其稳定性、重复性、回收率等。
方法:采用气相色谱法测定,色谱柱为30m.AC-1石英毛细管柱,检测器温度为230℃,以联苯胺为内标。
结果:线性范围0.5~2.5mg/mL,r=0.9993;回收率为99.4%,RSD=0.74%。
结论:采用GC法测定,方法简便、精确、重现性好,可很好地控制盐酸左旋咪唑的内在质量。
关键词:盐酸左旋咪唑;气相色谱;含量测定复方鹧鸪菜散为驱虫药物,用于小儿蛔虫病。
其含量测定方法,部颁标准采用的是紫外分光光度法。
该方法较繁琐,且在实验过程中大量使用三氯甲烷。
本篇介绍采用气相色谱法将盐酸左旋咪唑分离并测定其含量,经检验,该方法简便,稳定性好。
1 仪器与试药气相色谱仪:岛津GC-14C;氢火焰离子化检测器(FID):HW色谱工作站。
盐酸左旋咪唑对照品(中国药品生物制品检定所);复方鹧鸪菜散(市售,0.3g/包):由广东宏兴制药厂提供(批号为080411、080512、080613);联苯胺(化学原料药,含量大于98%)。
所有化学试剂均为分析纯(广州化学制剂厂),水为重蒸馏水。
2 方法和结果2.1 色谱条件色谱柱为30m.AC-1石英毛细管柱;检测器:氢火焰离子化检测器FID,氢气流速为50mL/min,空气流速为500mL/min;进样口温度为250℃,检测器温度为230℃,载气为高纯氮,标流量为0.1mL/min,分流比1/20,进样量为2μL,柱初始温度为230℃,停留10min,以10℃/min速度一阶升温到250℃。
2.2 内标溶液的配制在选定的色谱条件下,筛选试验结果表明,取联苯胺适量批号为(080203),加乙醇溶解并稀释成10mg/mL的溶液,作为内标较为理想。
2.3 对照品溶液的配制取盐酸左旋咪唑适量,精密称定,加乙醇溶解并稀释成5mg/mL的溶液,摇匀,即得。
HPLC法测定盐酸左旋咪唑片的含量研究

doi:10.11751/ISSN.1002-1280.2019.08.08HPLC法测定盐酸左旋咪唑片的含量研究周芷锦,林仙军,王彬,陈晓林(浙江省兽药饲料监察所,杭州311101)[收稿日期]2019-06-04㊀[文献标识码]A㊀[文章编号]1002-1280(2019)08-0051-05㊀[中图分类号]S859.79[摘㊀要]㊀建立盐酸左旋咪唑片中盐酸左旋咪唑含量测定的高效液相色谱法㊂采用AgilentEclipseXDB-C18(250mmˑ4.6mm),5μm色谱柱,Agilent1260DAD检测器,流动相为0.05mol/L磷酸二氢钾溶液(三乙胺调节pH值至7.0)-乙腈(80ʒ20,v/v);进样量为20μL;柱温为30ħ;流速为1.0mL/min;检测波长为214nm㊂结果表明:盐酸左旋咪唑在1.024 102.398μg/mL范围内线性关系良好(r=1.0000),高㊁中㊁低浓度回收率分别为97.4%㊁99.7%㊁99.3%㊂该方法定性㊁定量准确,适用于盐酸左旋咪唑片中盐酸左旋咪唑的含量测定㊂[关键词]㊀盐酸左旋咪唑片;高效液相色谱法;二极管阵列检测器作者简介:周芷锦,从事兽药检测相关工作㊂E-mail:miya_zt@163.comDeterminationofLevamisoleHydrochlorideinLevamisoleHydrochlorideTabletsbyHPLCZHOUZhi-jin,LINXian-jun,WANGBin,CHENXiao-lin(ZhejiangProvinceSupervisoryInstituteofVeterinaryDrugandFeed,Hangzhou311101,China)Abstract:ToestablishamethodforthedeterminationofLevamisoleHydrochlorideinLevamisoleHydrochlorideTabletsbyHPLC.AgilentEclipseXDB-C18(250mmˑ4.6mm),5μmcolumnwasused.Themobilephasewaspotassiumdihydrogenphosphatephosphoric(0.05mol/L,pHvalueadjustedto7.0withtriethylamine)-acetonitrile(80ʒ20,v:v),theinjectionvolumewas20μL,thecolumntemperaturewas30ħ,theflowratewas1.0mL/minanddetectedatthewavelengthof214nm.Theresultindicatedthatagoodlinearrelationshipwasacceptedintherangeof1.024 102.398μg/mL(r=1.0000).andtherecoveryofhigh,medium,low-Concentrationwere97.4%,99.7%,99.3%.Themethodisaccurateinqualitativeandquantitativeanalysis,andsuitableforthedeterminationofLevamisoleHydrochlorideTablets.Keywords:LevamisoleHydrochlorideTablets;highperformanceliquidchromatography;diodearraydetector㊀㊀左旋咪唑是一种广谱驱虫药,广泛应用于兽医临床治疗畜禽胃肠道线虫病㊁肺丝虫病和猪肾虫病当中㊂除作为驱虫药之外,盐酸左旋咪唑还具有免疫调节作用,是一种免疫增强剂,能提高巨噬细胞的吞噬能力,增强机体的体液免疫功能和细胞免疫功能[1]㊂研究发现,盐酸左旋咪唑可显著提高脾脏指数和法氏囊指数,对雏鸡有显著的增重效果,在一定程度上提高生长性能[2]㊂目前兽医使用中常见的剂型包括盐酸左旋咪唑片㊁盐酸左旋咪唑注射液㊂目前盐酸左旋咪唑片在‘中国兽药典(2015年版)“一部[3],‘美国药典“(USP40-NF35)[4]中均有收入㊂‘中国兽药典(2015年版)“一部[3]中,含量测定标准采用氯仿提取,高氯酸滴定的方法㊂该方法主要存在氯仿毒性较大,且过程较为繁琐,容易出现误差较大等问题㊂‘美国药典“[4]采用高效液相色谱法,但对实验室仪器设备有较高的要求㊂基于文献参考[4]-[6],本着优化简化实验条件和方法的目的,本文探讨用高效液相色谱法测定盐酸左旋咪唑片中盐酸左旋咪唑含量的方法研究㊂1㊀仪器与材料1.1㊀仪器㊀高效液相色谱仪,Angilent1260,配AngilentDAD检测器;METTLERTOLEDOXS205电子天平;KQ-500E型超声波清洗器(昆山市超声仪器有限公司);METTLERTOLEDO320酸度计㊂1.2试剂㊀乙腈㊁甲醇均为色谱纯,德国默克公司;磷酸二氢钾㊁磷酸㊁三乙胺均为分析纯;水为超纯水㊂盐酸左旋咪唑对照品来源为中国食品药品检定研究院,批号100167-201203,含量99.9%㊂1.3㊀样品㊀实验样品由杭州新港动物药业有限公司等四家单位提供,4个批次25mg规格的样品批号分别为:20190101,180529,190122,20180703;2个批次50mg规格的样品批号分别为:170901,190301㊂2㊀方法与结果2.1㊀溶液的配制2.1.1㊀对照品溶液配制㊀取盐酸左旋咪唑对照品约10mg,精密称定,置200mL容量瓶中,加入适量流动相溶解,并用流动相稀释至刻度,摇匀㊂2.1.2㊀供试品溶液配制㊀取本品20片,精密称定,研细,精密称取适量(约相当于盐酸左旋咪唑50mg),置100mL容量瓶中,加入流动相适量,超声10min溶解,用流动相稀释至刻度,摇匀;过滤,精密量取续滤液5mL,置50mL容量瓶中,用流动相定容,摇匀㊂2.2㊀色谱条件及系统适应性试验㊀采用十八烷基硅烷键合硅胶色谱柱(AgilentEclipseXDB-C18,4.6mmˑ250mm,5μm),流动相为0.05mol/L磷酸二氢钾溶液(三乙胺调节pH值至7.0)-乙腈(80ʒ20,v/v);进样量:20μL;柱温:30ħ;流速:1.0mL/min;用二极管阵列检测器在190 400nm范围内进行扫描,记录214nm波长处的色谱图㊂盐酸左旋咪唑对照品光谱图(图1),盐酸左旋咪唑对照品溶液色谱图(图2),供试品溶液色谱图(图3)㊂图1㊀盐酸左旋咪唑对照品溶液光谱图Fig1㊀SpectrogramofLevamisoleHydrochloridereferencesubstance图2㊀盐酸左旋咪唑对照品溶液色谱图Fig2㊀SolutionchromatogramofLevamisoleHydrochloridereferencesubstance图3㊀供试品溶液色谱图Fig3㊀Solutionchromatogramofthesample2.3㊀线性关系考察㊀取盐酸左旋咪唑对照品,精密称定,加流动相制成100μg/mL的溶液,并配制成1㊁2.5㊁5㊁10㊁25㊁50㊁100μg/mL等一系列浓度的对照品溶液㊂分别精密吸取20μL,注入色谱仪,测定㊂以对照品浓度(X)为横坐标,峰面积值(Y)为纵坐标进行线性回归,绘制标准曲线㊂在1.024 102.398μg/mL范围内线性关系良好,线性回归方程为Y=89.715X+14.765,线性相关系数为1.0000㊂2.4㊀回收率试验㊀以批号190301的样品作为本底样品,精密称取9份,每三份为一组,称取量为应称取样品量的一半(约相当于盐酸左旋咪唑25mg),置100mL容量瓶中;另精密称取盐酸左旋咪唑对照品,高㊁中㊁低浓度对照品加入量按照应称取量的80%㊁100%㊁120%的比例加入上述的三组100mL容量瓶中,并按照2.1.2的方法配制回收率试验用溶液㊂按照2.2色谱条件,精密量取20μL,注入色谱仪中,记录色谱图㊂计算回收率,回收率=(实测量/加入量)ˑ100%,实测添加量=测得总量-本底盐酸左旋咪唑量㊂回收率结果如表1所示,平均回收率为97.4%㊁99.7%㊁99.3%,说明检测方法的准确度良好㊂表1㊀回收率结果表Tab1㊀Resultsofrecoverytate添加浓度本底称样量/g本底含量/mg对照品添加量/mg测得总量/mg回收率/%平均回收率/%RSD/%80%0.101024.8315.3539.7797.30.102425.1715.5240.1696.50.101324.9015.4440.0898.397.40.9100%0.101825.0325.0849.8298.80.101925.0524.9850.0299.90.101524.9524.9349.97100.499.70.8120%0.101725.0034.7259.4499.20.102325.1535.2159.9198.70.101624.9834.9759.9099.999.30.62.5㊀精密度㊀取回收率试验的9份样品测定,RSD值按照高㊁中㊁低浓度三组分别为0.9%,0.8%,0.6%,说明精密度良好㊂2.6㊀稳定性试验㊀取重复性试验的供试品溶液,在0,1.5,3,6,12,16h时间点进样,测得器峰面积分别为4483.87744,4474.66260,4492.76318,4482.00439,4516.70264,4501.69922,其检测结果的RSD为0.3%,说明在16h内供试品溶液基本稳定㊂2.7㊀耐用性试验㊀采用三台不同的液相和三根不同品牌的色谱柱分三组对批号为20190101的样品进行含量测定㊂A组为Agilent1260DAD液相色谱仪,配AgilentC18(4.6mmˑ250mm,5μm)色谱柱;B组为Agilent1260VWD液相色谱仪,配WatersC18(4.6mmˑ250mm,5μm)色谱柱;C组为Waters2695-2487液相色谱仪,配AgilentC18(4.6mmˑ150mm,5μm)液相色谱柱㊂样品配制方法如2.1.2,色谱条件见2.2,含量测定结果如表2所示,同一样品三组含量测定结果分别为100.5%,101.3%,101.2%,RSD值为0.5%,说明该检测方法对液相和色谱柱品牌无特殊要求,耐用性良好㊂2.8㊀实际样品含量测定㊀取1.3中的所有样品,每个样品平行3次,依法测定其含量,结果见表3㊂表2㊀不同液相与不同品牌色谱柱的含量测定比较Tab2㊀ComparisonofcontentdeterminationofdifferentHPLCanddifferentbrandcolumns组别称样量/g含量/%平均含量/%组内RSD/%组间RSD/%ABC0.1813100.590.1821100.140.1812100.630.1813100.720.1821101.440.1812101.730.1813101.060.1821101.080.1812101.60100.50.3101.30.5101.20.30.5表3㊀液相测定结果Tab3㊀DeterminationresultsofHPLC批号规格/mg称样量/g含量/%平均含量/%RSD/%20190101250.1813100.590.1821100.140.1812100.63100.50.3180529250.1371100.140.1345100.270.1341100.32100.20.1190122250.228999.570.2295100.160.2292100.0699.90.320180703250.2237106.310.2256106.290.2250104.75105.80.8170901500.2269107.650.2264106.870.2264106.13106.90.7190301500.2042102.260.2038102.010.2041101.22101.80.53㊀讨论与结论3.1㊀与容量分析结果比较㊀取6批次样品,按照‘中国兽药典(2015年版)“一部中盐酸左旋咪唑片的含量测定项下的滴定法进行测定,结果见表4,并与高效液相色谱法比较,两者相对偏差均不超过1.0%,结果无显著差异㊂但滴定法存在以下问题:一是需使用毒性较大的三氯甲烷进行提取;二是步骤过长过多,实验误差较大;三是终点蓝色难以判断,容易过终点误读,导致实验结果偏高㊂相比较而言,高效液相色谱法更简便,也无需三氯甲烷㊁醋酐等危险化学试剂,更有利于实验的开展,实验结果也更为准确可靠㊂表4㊀液相含量测定与容量分析结果比较Tab4㊀ComparisonsbetweenHPLCandvolumetricanalysis样品批号规格/mg液相含量/%滴定含量/%相对偏差/%2019010125100.5101.00.218052925100.2100.2ɤ0.051901222599.998.20.92018070325105.8105.00.417090150106.9107.60.319030150101.8102.50.33.1㊀提取方式的选择㊀在提取溶剂选择上,分别采用了65%甲醇㊁甲醇和流动相三种溶剂进行超声提取㊂结果表明纯甲醇提取后样品峰形较差,65%甲醇和流动相提取后的样品峰形较好,本文采用了流动相提取㊂在是否过滤的选择上,过滤与不过滤峰面积相对偏差为0.2%,对含量测定影响不大,由于过滤后溶液更澄清,更有利于第二步配制,本文采用了过滤的方法㊂在超声时间的选择上,实验过程中分别采用了超声5㊁10㊁20min三种方式,峰面积分别为5255.33350,5262.27686,5247.13623,RSD为0.1%,因此选择超声时间为10min㊂3.2㊀检测波长的选择㊀对盐酸左旋咪唑溶液在190 400nm波长范围内扫描,扫描结果表明在212nm波长处有最大吸收,结合文献[5]-[6]报道中基本选择214nm为检测波长,基于212和214nm属于光谱扫描误差内,因此还是选择214nm为检测波长,与文献保持一致㊂3.3㊀流动相的选择㊀本文采用了甲醇-0.2%磷酸溶液以及乙腈-0.05mol/L磷酸二氢钾溶液两种流动相配比进行试验,供试品溶液采用流动相进行稀释配制成50μg/mL的溶液㊂结果表明,甲醇-0.2%磷酸溶液流动相下的盐酸左旋咪唑峰峰形不理想,峰形有较明显的拖尾现象,且有较明显的溶剂吸收峰,出峰时间在8min左右,理论板数在8000左右㊂而乙腈-0.05mol/L磷酸二氢钾溶液(三乙胺调节pH值至7.0)(20ʒ80,v/v)的流动相下的盐酸左旋咪唑峰峰形对称性良好,无干扰,无拖尾,出峰时间在13min左右更为合理,无明显的溶剂吸收峰,理论板数在10000以上㊂因此选择后者作为流动相更适宜㊂采用高效液相色谱法对盐酸左旋咪唑片中盐酸左旋咪唑的含量进行测定,对提取溶剂,提取方式和色谱条件进行了研究和优化,方法学验证结果表明该方法定量准确,与现行有效标准的含量结果一致,为盐酸左旋咪唑片的质量控制提供了更快捷㊁便利的操作方法㊂参考文献:[1]㊀林燕飞,张琴,高芳芳.盐酸左旋咪唑对免疫低下小鼠免疫功能的影响.[J].食品与药物,2018,20(6):460-464.LinYF,ZhangQ,GaoFF.Determinationofrheologicalproper⁃tiesofbutenafinehydrochloridegelbyrotationalrheology[J].ChineseJournalofFoodandDrug,2018,20(6):460-464.[2]㊀郭洋,刘敏跃,李鹏,等.盐酸左旋咪唑对雏鸡免疫功能及生长性能影响的研究[J].饲料工业,2015,36(10):10-12.GuoY,LiuMY,LiP,etal.Effectsoflevamisoleonimmunologi⁃calfunctionandgrowthperformanceinchickens[J].ChineseJournalofFeedIndustry,2015,36(10):10-12.[3]㊀中国兽药典委员会.中华人民共和国兽药典.2015年版一部[S].ChineseVeterinaryPharmacopoeiaCommittee.ChineseVeterinaryPharmacopoeia,volume1,2015Edition[S].[4]㊀美国药典40.[S].2017USP40-NF35[S].2017.[5]㊀刘素梅,李华岑,韩立,等.万乳康中盐酸左旋咪唑含量测定方法的研究[J].中国兽药杂志,2010,44(2):26-28.LiuSM,LiHC,HanL,etal.Determinationoflevamisolehydro⁃chlorideinWanRukang[J].ChineseJournalofVeterinaryDrug,2010,44(2):26-28.[6]㊀李忠红,张玫,樊夏雷,等.三国药典盐酸左旋咪唑片含量测定方法比较[J].中国药事,2011,25(5):497-499.LiZH,ZhangM,FanXL,etal.Comparisonofdeterminationmethodsoflevamisoletabletsinthreepharmacopoeias[J].JournalofChinesePharmaceuticalAffair,2011,25(5):497-499.(编辑:陈希)。
HPLC法检测保健品口服液中的盐酸左旋咪唑

HPLC法检测保健品口服液中的盐酸左旋咪唑李晓茵;韦林;庄少珊;黄丽芹;吴淋富;郑晓渲;李聪聪【期刊名称】《生物化工》【年(卷),期】2024(10)1【摘要】目的:建立高效液相色谱法检测保健品口服液中盐酸左旋咪唑的方法。
方法:采用C18-WR色谱柱(4.6 mm×250 mm,5μm),流动相为乙腈-0.05 mol/L磷酸二氢钾溶液(用三乙胺调节pH=7)(25∶75,体积比),流速1.0 mL/min,检测波长210 nm,柱温30℃,进样量10μL。
结果:盐酸左旋咪唑在8.634~86.340μg/mL时与峰面积线性关系良好(R=0.9997);低、中、高加标量对应的平均回收率分别为99.7%、99.2%和100.3%,RSD分别为1.40%、0.64%和0.78%,检测限为0.4317μg/mL。
随机对市售16批样品进行检测,均未检出盐酸左旋咪唑。
结论:该方法简便、快速、准确,可用于保健品口服液中非法添加盐酸左旋咪唑的检测。
【总页数】4页(P40-43)【作者】李晓茵;韦林;庄少珊;黄丽芹;吴淋富;郑晓渲;李聪聪【作者单位】汕尾市食品药品检验所;汕尾市高级技工学校;广东药科大学【正文语种】中文【中图分类】O657.7【相关文献】1.胶体金免疫层析试纸条法和HPLC-QTOF-MS法检测保健品中那非类化学药物的比较2.HPLC法检测保健品中违禁添加9种化学降糖药物3.HPLC-DAD 法快速筛查检测中成药及保健品中违法添加双胍类化学降糖药物分析4.HPLC-DAD法检测盐酸左旋咪唑注射液中左旋咪唑的含量5.RP-HPLC法检测甜梦口服液(甜梦合剂)中淫羊藿苷、紫丁香苷和23-乙酰泽泻醇B的含量因版权原因,仅展示原文概要,查看原文内容请购买。
HPLC-DAD法检测盐酸左旋咪唑注射液中左旋咪唑的含量

科研动态48 中国动物保健 | 2018.12HPLC-DAD 法检测盐酸左旋咪唑注射液中左旋咪唑的含量*林仙军1,章荣叶2,周芷锦1,汤赛飞3,王彬1,陈晓林1,陆春波1(1.浙江省兽药饲料监察所 杭州 311101;2.台州市屠宰管理所 浙江台州 318000;3.浙江建安检测研究院有限公司 杭州 310006)摘要:本研究建立了HPLC-DAD法测定盐酸左旋咪唑注射液中左旋咪唑含量的方法。
采用Agilent Eclipse XDB-C18(250 mm×4.6 mm,5µm)色谱柱,流动相为0.05mol/L磷酸二氢钾溶液-乙腈(80∶20,v/v),流速为1.0mL/min,进样量为10μL,柱温为30℃,用二极管阵列检测器在190~400nm扫描,记录212nm波长处的色谱图。
左旋咪唑在1~500μg/mL浓度范围内线性良好,相关系数为0.999 99。
在2.5、5.0和10.0mg/g三个添加浓度下,加样回收率为99.1%~100.5%,RSD≤1.5%,检测限为2.5mg/g。
本方法简便快捷,准确度和灵敏度高,可用于左旋咪唑注射液的质量控制。
关键词:左旋咪唑;高效液相色谱;含量测定;二极管阵列检测器盐酸左旋咪唑是广谱驱虫药和生物反应调节药[1],对牛、羊、猪、犬、猫和禽的胃肠道线虫、肺线虫及猪肾虫病具有良好驱虫活性[2-3]。
盐酸左旋咪唑注射液质量标准收载于《中国兽药典》2015年版一部[4],标准采用三氯甲烷萃取、高氯酸滴定法(非水测定法)进行含量测定,方法操作繁琐、费时,高氯酸滴定液能与多种药物反应,专属性不高。
检测中所用试剂三氯甲烷、醋酐等对人体有危害性,对环境污染也较严重。
目前文献报道测定盐酸左旋咪唑含量的方法除高氯酸滴定法外,主要以高效液相色谱法为主,有测定饲料[5]、绵羊血浆[6]、猪鸡组织[7]、盐酸左旋咪唑粉[8]、复方盐酸左旋咪唑注射液[9]和万乳康[10]等各类样品中盐酸左旋咪唑的含量,以及盐酸左旋咪唑原料中的有关物质[11],但目前尚未见高效液相色谱法(HPLC)测定盐酸左旋咪唑注射液中左旋咪唑含量的报道。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
验证性试验
实验八 盐酸左旋咪唑片的含量测定
一、实验目的
1.掌握液-液提取分离方法及其操作。
2.掌握非水酸碱滴定法测定弱碱性药物的原理。
二、仪器与试剂
1.仪器
Mettler AL204电子天平 分液漏斗 规格:125mL
研钵 定量滤纸(直径10cm ) 刻度移液管 规格:5mL
胖肚移液管 规格:25mL 50 mL 酸式滴定管 规格:10mL
2.试剂
盐酸左旋咪唑片 规格:25mg/片
氢氧化钠 二氯甲烷 结晶紫指示液 高氯酸滴定液 冰醋酸 蒸馏水
三、实验原理
盐酸左旋咪唑原料:以有机碱盐(BH +)的形式存在,滴定过程就是一个置换滴定,即强酸滴定液置换出与游离碱结合较弱的酸。
BH +.A - + HClO 4
BH +.ClO 4-
+ HA 盐酸左旋咪唑制剂:左旋咪唑以游离有机碱(B )的形式存在,为弱碱,溶于冰醋酸后,其碱强度被均化到溶剂阴离子的强度水平,增加左旋咪唑的碱性,以结晶紫为指示剂,可用高氯酸滴定液进行滴定。
BH + + OH -
B + H 2O
B + HA C
BH + + A C - HClO 4 + HA C
ClO 4- + H 2A C + H 2A C + + A C
- 2HA C
四、实验内容
盐酸左旋咪唑片
yansuan Zuoxuanmizuo Pian
Levamisole Hydrochloride Tablets
C 11H 12N 2S •HCl 240.76
本品含盐酸左旋咪唑(C 11H 12N 2S •HCl )应为标示量的90.0%~110.0%。
[含量测定] 取本品20片,精密称定,研细,精密称取适量(约相当于盐酸左旋咪唑0.2g ),置分液漏斗中,加水10mL ,振摇使盐酸左旋咪唑溶解,加氢氧化钠试液5mL ,稍振摇后,精密加入二氯甲烷50.0mL ,振摇提取,静置,分层后分取二氯甲烷液,经干燥滤纸滤过,弃去初滤液,精密量取续滤液25mL ,加冰醋酸15mL 与结晶紫指示液1滴,用高氯酸液(0.1mol/L )滴定,至溶液显蓝色,并将滴定结果用空白试验校正即得。
每1mL 高氯酸液(0.1mol/L)相当于24.08mg 的C 11H 12N 2S •HCl 。
.HCl
计算:盐酸左旋咪唑标示量% =0V V F T D W 100%W ⨯⨯⨯⨯⨯⨯(—)标示量
V O :空白试验消耗高氯酸滴定液的体积(mL );
V:供试品消耗高氯酸滴定液的体积(mL );
F :高氯酸滴定液浓度校正因数; T :滴定度;
W :平均片重(g ); W: 供试品片粉取样量(g );
D:稀释倍数。
五、注意事项
1.注意滴定液浓度及其标定时的温度,若测定样品时温度较标定时高或低2~10℃,则应以冰醋酸体积膨胀系数0.0011校正。
2.所用试剂含水量均应在0.2%以下,冰醋酸在使用前应做空白试验:取冰醋酸5~10mL 于50mL 锥形瓶中,加结晶紫指示剂1滴,应为紫色,加高氯酸液(0.1mol/L )1滴(0.01mL ),即应变为黄绿色,若为蓝色,则表明有水分存在,可加醋酐脱水,或加醋酐后重蒸一次。
3.冰醋酸中绝大部分分子是呈氢键缔合成环状的二聚物,沸点高(118℃),但冰醋酸具挥发性,故:高氯酸应密闭贮存,滴定液装入滴定管后,其上宜用一干燥小烧杯罩上,最好用自动滴定管进行滴定。
4.本法滴定终点的颜色变化复杂,重点判定以电位法为准,同时采用指示液对照观察终点颜色的变化。
一般非水滴定法终点指示剂颜色的判定:应根据电位滴定来校正,决不可认为终点颜色与空白一致即可。
5.冰醋酸有刺激性;高氯酸与有机物接触、遇热易引起爆炸,应用时需注意安全;结晶紫指示液不能放置过久。
六、思考题
1.二氯甲烷为什么用干燥滤纸过滤?
2.盐酸左旋咪唑片为什么不能用高氯酸直接滴定?
3.配制0.1mol/L 高氯酸液1000mL,加高氯酸(70%,d=1.75)8.5mL,则为除去8.5 mL 高氯酸中的水分,应加入相对密度为1.08,含量为97.0%的醋酐多少mL ?
七、参考文献
《中国药典》2010年版二部,476,化学工业出版社。