分子系统发育分析5
系统发育生物学

系统发育生物学是一门研究生物多样性演化,以及生物间的亲缘关系的学科。
它利用综合生态和遗传学方法,研究各个物种之间的差异和相似性,通过分类学的方法将物种进行分类,并推断每个物种之间的演化历史。
本文将为您介绍的基本概念、应用以及近年来的研究进展。
一、基本概念系统发育学主要研究一个物种或者群体的亲缘关系,也就是它们之间的演化关系。
演化学家在研究物种时,可以注意到它们的功能、形态、行为、遗传和地理分布等方面的变异。
通过对遗传基因的分析,我们可以发现更多物种间的关系。
我们会把采用获得更多信息的研究方法进行分类的方式称为分类学。
这种分类学可以被看作是的一种分支。
在中使用的关键方法是比较形态学和分子遗传学。
比较形态学是基于动植物的外形、骨骼、体重、眼睛和器官等性状的比较。
而分子遗传学在分析DNA、RNA 或蛋白质序列构成的基础上,来揭示物种间的亲缘关系。
分子系统发育是目前使用最广泛的系统发育分析方法之一。
二、应用系统发育学在分类与物种定义、生物多样性保护以及人类相关疾病的研究等方面,具有广泛的应用价值。
下面我们将一一介绍。
1. 分类与物种定义物种定义是根据生物体生物学特点、四合一标准、分子分析等多种分类方法来进行的。
采用基于比较形态学和分子遗传学的方法来确定物种的医学生物学意义,不仅可以识别并分离不同的物种,还可以推测不同物种的分化历史。
2. 生物多样性保护生物多样性是指生物体的不同种类、品种和群体的总和。
而生物多样性保护是通过保护和管理这些生物之间的相互关系,保护整个生态系统。
例如,生物多样性研究可以识别需要特别保护、受威胁的物种,以及需要激励的营造性征。
通过生物多样性研究,可以从宏观和微观角度深入研究生态演化、特定群体的分化、物种多样性等,并从中发掘解决生态学、技术和经济等多个领域存在的问题的方法。
3. 人类相关疾病的研究在人类相关疾病的研究中,以及相关的进化生物学方法被应用。
这些方法可以揭示人类与其它物种的亲缘关系,从而推测疾病和我们的演化历程有关系。
生物信息基础 第6章 系统发育分析

生 物 信 息 基 础 - Basics in Bioinformatics 模式识别与智能系统实验室 5
•• 例2:冠状病毒全
基因组核酸序列 的系统发育树
[1] Peter Forster et al., Phylogenetic network analysis of SARS - CoV - 2 genomes, PNAS 2020.
表型特征
• 基因组数据方面的差异
– 数据丰富 – 建立了严格的数学模型
基因型特征
生 物 信 息 基 础 - Basics in Bioinformatics 模式识别与智能系统实验室
7
表型特征的局限性
• 表型特征的局限性
– 趋同进化的影响(表型相似并不总反映基因相似)
• 人、软体动物、蝗虫
– 难以选择合适的表型特征
叶结点排列整齐,内部结点 可以反映进化时间的顺序
分枝长度与物种/序列的进 化时间成正比
两种树都可在分枝上标注信息(分支长度、进化时间以及 其它数值)
生 物 信 息 基 础 - Basics in Bioinformatics 模式识别与智能系统实验室
12
2. 叉树 (内部结点的分叉)
二歧分叉
2G I
•
计算方法
优化算法
聚类算法
简约法(MP) Parsimony
最 大 似 然 法 (ML) MaximumLikelihood
数据类型 距离数据 特征数据
进化距离最小二乘法
UPGMA法
邻 接 法 (NJ) NeighborJoining
生 物 信 息 基 础 - Basics in Bioinformatics 模式识别与智能系统实验室
系统发育分析
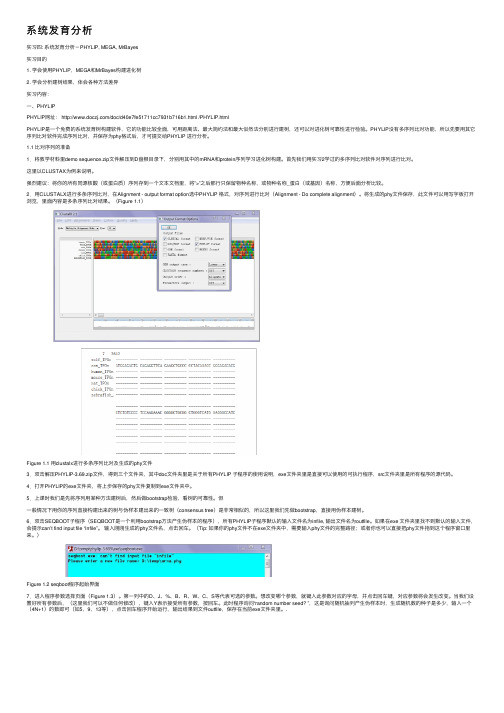
系统发育分析实习四: 系统发育分析-PHYLIP, MEGA, MrBayes实习⽬的1. 学会使⽤PHYLIP,MEGA和MrBayes构建进化树2. 学会分析建树结果,体会各种⽅法差异实习内容:⼀、PHYLIPPHYLIP⽹址: /doc/d40e7fe51711cc7931b716b1.html /PHYLIP.htmlPHYLIP是⼀个免费的系统发育树构建软件,它的功能⽐较全⾯,可⽤距离法、最⼤简约法和最⼤似然法分别进⾏建树,还可以对进化树可靠性进⾏检验。
PHYLIP没有多序列⽐对功能,所以先要⽤其它序列⽐对软件完成序列⽐对,并保存为phy格式后,才可提交给PHYLIP 进⾏分析。
1.1 ⽐对序列的准备1.将教学材料⾥demo sequence.zip⽂件解压到D盘根⽬录下,分别⽤其中的mRNA和protein序列学习进化树构建。
⾸先我们⽤实习2学过的多序列⽐对软件对序列进⾏⽐对。
这⾥以CLUSTAX为例来说明。
强烈建议:将你的所有同源核酸(或蛋⽩质)序列存到⼀个⽂本⽂档⾥,将”>”之后那⾏只保留物种名称,或物种名称_蛋⽩(或基因)名称,⽅便后⾯分析⽐较。
2.⽤CLUSTALX进⾏多条序列⽐对,在Alignment - output format option选中PHYLIP 格式,对序列进⾏⽐对(Alignment - Do complete alignment)。
将⽣成的phy⽂件保存,此⽂件可以⽤写字板打开浏览,⾥⾯内容是多条序列⽐对结果。
(Figure 1.1)Figure 1.1 ⽤clustalx进⾏多条序列⽐对及⽣成的phy⽂件3.双击解压PHYLIP-3.69.zip⽂件,得到三个⽂件夹,其中doc⽂件夹⾥是关于所有PHYLIP ⼦程序的使⽤说明,exe⽂件夹⾥是直接可以使⽤的可执⾏程序,src⽂件夹⾥是所有程序的源代码。
4.打开PHYLIP的exe⽂件夹,将上步保存的phy⽂件复制到exe⽂件夹中。
5种蚌螨ITS2基因片段序列及其系统发育分析

Is 因序 列长 度 为 28b ( gp , 中保 守位 T 2基 5 p 含 a ) 其 点 16个 , 8 可变 位点 5 7个 ( 2 , 变异 率达 到 2 . 表 )总 2
1 , 间变 异 率 在 7 4 ~1 . % 之 间 ; % 种 .% 49 转换 位 点 1 个 , 换位 点 l 1 颠 8个 , 换 位 点 低 于 颠 换 位 点 , 转 转
换 颠换 比值 为 06 ; G、 T4种 碱基 的平 均 含 量 .1A、 C、
分 别 为 2 . % 、7 2 、8 1 、5 2 ; 均 嘧 啶 9 5 1 . % 1. % 3 . % 平
A 7 73 ) 使 用 P U 4 O l 件 中 的 最 大 似 然 Y 0 20 , A P . b0软 法( ) ML 和邻 接 法 ( J 构 建 系 统 树 , 对 系统 树 采 N) 并
毛序 的微 小变 化加 以区分 , 对 判定 一 些 蚌螨 这
厚蚌螨 ( c sps 采 集 于 南 昌 市 郊 池 塘 , r s e) ai 其
近 似种 的分类 地 位 产 生 了影 响 。 因此 , 子 标记 技 分 术 也用 于对蚌 螨 的近似种 与 隐蔽种 的鉴定 ¨ J 卜 。
1 2 实验方 法 .
该区段适合用来 分析动物亲缘 关系较 近 的种 、 种下分
类 阶元 以及地 理种群之 间的系统关系 。 近几 十年来 , 国内外 已有许 多使 用 IS T 2序列 研
12 1 基 因组 D A的提 取 .. N
每种蚌 螨 取 1只个 体
的 研 磨 缓 管 中 , 入 3 加 0
用 10 0次 b o t p检 验分 析 。 0 ot r sa
浅谈系统发育分析方法

分 子 生 物 学 技 术 的 发 展 以 及 生 物 分 子 数 据 的 积累 ,系统发 育分析进 入 了分 子层次 。 分子 系统发 育分 析中常用的生物大分子是 作 为生 命机 器的蛋白质和作为遗传物质的 核酸 。早期 的研 究工作主要是 利用不同物 种 中同一种 基因 /蛋 白质序列 的异 同重建 系统 发育树 ,并研 究各物种 的进化关 系 。 近年来 ,较多模式生 物基 因组测序 任务的 完 成 以 及 蛋 白质 组 学 的发 展 ,为 从 “组 ”水 平 进 行 系 统 发 育 研 究 提 供 了条 件 ,但 同 时也对现有的 系统发 育分析 方法提 出了挑 战 。 源自l§I曩 ◇ 0≮ ◇≮器 毒羹 一
系统 发育 分析 ; 分 子进 化 ; 序 列 比 对
引言
地 球上 的一 切生 命形 式 ,不 管是 现 存 的还 是 已 经 灭 绝 了的 ,都 由于 一 个 共 同 的进化 历史而有着不 同程度的关联 ,这种 关联也使研究物种之 间进化关 系的学科一 系统 发育学 变得非常有意 义。追溯 生物 界 不同生物类型的起源及进化关系 ,即重 建 生 物 类 群 的 系 统 发 育 树 已经 成 为 生 物 信 息 学中一个十分重要的研究内容 ,并 日益受 到 广 泛 的 关 注 。
分子进化分析讲解

—— 寻找这棵正确的树
+ 分子进化分析介绍 + 系统发育树重建方法 + 常用分子进化与系统发育分析的软件
选择数据(核酸/蛋白质,外围支) 多序列比对(自动比对,手工比对)
选择建树方法及取代模型 建立进化树 进化树评估
+ 从多重序列比对到构建进化树有多种算法, 可分两大类:
+ 基于距离的方法
– Tree 1长4,Tree 2& 3长2
+ 同理,综合所有信息位点:
– Tree 1长4,Tree 2长5,Tree 3长6
+ 计算结果:MP tree的最优结果为Tree 1
+ 又称距离矩阵法,首先通过各个物种之间的 比较,根据一定的假设(进化距离模型)推 导得出分类群之间的进化距离,构建一个进 化距离矩阵。进化树的构建则是基于这个矩 阵中的进计化算距序离列关的距系离,建立距离矩阵
– 首先通过各个序列之间的比较,根据一定的假 设(进化距离模型)推导出分类群之间的进化 距离,构建一个进化距离矩阵。进化树的构建 则是基于这个矩阵中的进化距离。
+ 基于特征的方法
– 不计算序列之间的距离,而是将序列中有差异 的位点作为单独的特征,并依据这些特征来建
+ 基于距离的方法
– 非加权分组平均法(UPGMA) – 最小近乎距离(ME) – 邻近法(NJ)
真细菌 真核生物
古生菌
随着距非洲距离越来越长, 遗传多样性的衰退程度, 正好沿着人类早期迁徙的 路线慢慢增大。
53个人的线粒体基因组 (16,587bp)
非洲人相对其他大陆上的 人类在基因上极为多样化
人类迁移的路线
一、系统发育树(Phylogenetic tree)
分子系统学
分子系统学分子系统学是指通过对生物大分子(蛋白质、核酸等)的结构、功能等的进化研究,来阐明生物各类群(包括已绝灭的生物类群)间的谱系发生关系.相对于经典的形态系统分类研究,由于生物大分子本身就是遗传信息的载体,含有庞大的信息量,且趋同效应弱,因而其结论更具可比性和客观性.尤为重要的是,一些缺乏形态性状的生物类群(如微生物和某些低等动、植物)中,它几乎成为探讨其系统演化关系的唯一手段.由于分子系统学的上述特点,自其诞生之日起,就逐渐在各种生物类群的系统发生研究中得到了广泛的应用.总的说来,迄今分子系统学的研究所获得的生物类群间亲缘关系的结果,大多都和经典的形态系统树相吻合.但是,在一些生物进化谱系不明或模糊关键环节上,它得出的结果却往往和形态系统学的推测大相径庭.1研究步骤分子系统学研究的主要方法是根据分子生物学数据构建生物类群的谱系发生树.它一般包括以下程序:1.首先确定所要分析的生物类群,选择该类群中相关亚类群的一些代表种类;确定所要分析的目的生物大分子(包括DNA序列、蛋白质序列等)或它们的组合;2.设法获得它们的序列数据或其它相关数据(如限制性内切酶(I LP)、随机扩增多态DNA( )、DNA序列等),DNA序列的数据可以通过GenBank获得,也可以通过实验室的研究(设计特异引物进行PCR扩增和序列测定)而获得;3.对获得的相关数据进行比对(pairwisealignment)或其它的数学处理,如转变成遗传距离数据矩阵;通过一些遗传分析软件(常用的计算机软件如:PHYLIP J、PAI J、MEGA[J 等)对这些处理后的数据,并基于一定的反映DNA序列进化规律的数学模型构建分子系统树;4对构建的系统树做相应的数学统计分析以检验系统树的可靠性等.值得注意的是,在分析具体的研究对象时,上述各个环节是紧密联系的一个整体,要获得一个正确的结论,必须综合考虑每一环节之间的内在联系.比如目的基因的选择、数据处理和分析的分类群之间、构树方法和分析软件的选择之间都有密切的联系.2涉及议题基因树和物种树分子系统学的目的就是通过基因树来推测物种树.基因树是根据生物大分子的序列数据(主要为DNA序列数据)构建的谱系树,物种树则是反映物种实际种系发生的谱系树.人们期待着得到的基因树和物种树相一致,然而实际情况往往并非如此.Nei(1987)描绘了二种谱系树之间所有可能的关系,认为二种谱系树之间至少存在二个方面的差异:一是基因树的分化时间早于物种树,二是基因树的拓扑结构可能与物种树不一致(二个或多个基因树之间存在着差异)如何将由多个基因或基因组建立的基因树综合成一个物种树,是分子系统学面临的一个主要难题.Maddison(1997)认为:基因重复所导致的并源而非直源关系的产生,不同生物类群问基因的水平转移,系统演化分歧事件发生后产生的分子性状的多型性引起的谱系选择等生物学因素是造成二者不一致的主要原因.相应地,分子系统学研究中一定要选择直源基因而非并源基因,选择水平转移事件较少的树,采用基于大量独立进化的基因位点进行分析等等,都不失为一种行之有效的方法,更有利于获得一个可靠的树.分类群的选择分子系统学研究中如何选择所研究的对象——内类群的选择是一个非常值得注意的问题.内类群选择(内类群的数目及选择依据等)的科学性与否直接影响到所得结论的可靠性.关于内类群的数目,目前大多数分子系统学家认为,当所分析的序列长度一定时,尽量选择较多的分类群有助于获得更准确的结论,而内类群选择的依据主要体现在:(1)结合古生物学,形态学等各方面证据,尽量保证所选择的分类群确为一个单系发生的类群;(2)分类群的选择并非是随机的,尽量使其在所研究的生物类群中具有代表性;(3)在某些因具有明显长枝效应(或短枝效应)而导致的系统关系不确定的分支间增加分类群有助于减弱或消除这种效应.另外,在构建分子系统树中,同样需要选择外类群以确定系统发生树的基部位置,从而确定进化的方向.外类群的选择可以是单个(单一外类群),也可以是多个(复合外类群).在所研究的内类群数目不多且二者之间的极性关系十分确定的情况下,单个外类群足以说明问题.而在较为复杂的分析中,通常选择复合外类群以保证所得结论的可靠性[11].随机选择的外类群,极有可能因为亲缘关系较远,导致所得结果的不确定性增大.因此,在选择外类群时,必须结合其它分类学上的证据,或者在做详细的系统发育研究之前,首先对所研究的内、外群的关系进行初步探讨,以便于选择较为理想的外类群.最理想的外类群应该是该内群的姐妹群,因为二者间拥有较多的共近裔性状.目的基因的选择分子系统学研究中目的基因的选择也是一个至关重要的问题.一般来说,要根据所研究的具体分类群选择适宜的基因:在高级分类阶元(科级以上)间的系统发生分析中,选择一些在进化中较为保守的基因或基因片段(如核编码的蛋白质(酶)基因、核糖体基因(18S rRNA基因、28S rRNA基因)等);在较低级的分类阶元间,可以选择进化速率较快的基因或基因片断(如某些核编码基因的内含子或转录间隔区(ITS)以及一些细胞器基因(线粒体基因和叶绿体基因)等).当然,每一个具体的研究对象,可以选择的基因数目可以是多个的,至于哪些是最有效的,这通常要依据具体情况做比较分析后才能得出结论.条件允许的话,可以作多基因或多基因组合分析后寻求一致树来加以解决.有时针对某些涉及到多种层次分类阶元的复杂分类群时,还可以采取组合分析的方法:即推断位于系统树基部的深层次的谱系发生时,运用较保守的基因作为目的基因;推断位于系统树中段的谱系发生时,采用进化速率较为适中的基因;在系统树顶端的终端分类单元时,采用进化速率较快的基因.这样可以在不同阶层的演化关系中都获得可信的结果.基因序列数据的比对选择了适宜的目的基因并通过基因的扩增(PCR技术)和序列测定后,就获得了各个目标生物类群的DNA序列数据,对所获得的同源DNA序列进行比对是分析中的关键环节.所谓比对是指通过插入间隔(gaps)的方法,使不同长度的序列对齐达到长度一致,并确保序列中的同源位点都排列在同一位置.其中间隔的处理对后续的系统学分析有明显的影响.序列比对目前通常基于以下二种原理:点标(dot plot)法和记分距阵(scoring ma仃ix)法.基因树的构建方法目前,构建基因树的方法很多,常用的主要有二大类:距离法(distancemethod):是将序列数据转变成数据(遗传距离)矩阵,然后通过此数据矩阵构建系统树、具体性状法(dis—cretecharacter method):直接分析序列上每个核苷酸位点所提供的信息构建系统树,它又包括最大简约法(MP)和最大似然法以及由ML法延伸的贝叶斯法(Bayesianmetl-,od).距离法该方法基于这样一种假设,即只要获得一组同源序列间的进化距离(遗传距离),那么就可以重建这些序列的进化历史.距离法中以邻接法(NJ)最为常用.邻接法是由Saitou和Nei(1987)提出,其原理是逐步寻找新的近邻种类(序列),使最终生成的分子树的遗传距离总长度为最小.该法虽并不检验所有可能的拓扑结构,但在每阶段诸物种(序列)聚合时都要应用最小进化原理,故而被认为是ME的一种简化方法.最大简约法该方法源于形态学的分支系统学研究,而最早被Fitch(1971)用于核苷酸数据研究.它是一种最优化标准,遵循“奥卡姆剃刀(Ockharn’S razor)原理,即假设由一祖先位点替换为另一位点时,发生的替换数目最少的事件为最可能发生的事件.在实际应用中,由于MP法只考虑所谓的“信息位点”,所得的进化树是最短的、也是变化最少的进化树.因而,简约法的“最小核苷酸替换数目”原则也意味着“异源同型事件(homoplastic event)(即平行替换、趋同替换、同时替换和回复突变等)最少.最大似然法该法最早由Felsenstein(1981)提出,其原理是以一个特定的替代模型分析一组既定的序列数据,使获得的每一个拓扑结构的似然率均为最大,再挑出似然率值最大的拓扑结构作为最终树这里所分析的参数是每个拓扑结构的枝长,并对似然率的最大值来估算枝长.迄今的研究表明,在分类群数目较大、序列长度较长的复杂分析中,ML法的分析结果优于其它任何方法。
微生物的进化系统发育
03
生物信息学方法将有助于发现 新的进化规律和模式,为进化 生物学提供新的理论框架和见 解。
感谢您的观看
THANKS
微生物的进化关系分析
进化关系分析主要关注不同微生物种 群之间的遗传差异和相似性,通过比 较基因组学、蛋白质组学等方法来研 究。
VS
进化关系分析有助于揭示微生物种群 之间的亲缘关系和演化历程,对于理 解微生物多样性和生态系统的功能具 有重要意义。
微生物的进化速率和方向
进化速率是指பைடு நூலகம்种在进化过程中基因序列、形态特征等发生变化的速度,而进化方向则是指物种在进 化过程中所呈现的趋势或路径。
微生物的进化系统发育
目录
• 微生物的进化历程 • 系统发育学的基本概念 • 微生物的系统发育分析 • 微生物进化系统发育的应用 • 微生物进化系统发育的未来展望
01
微生物的进化历程
微生物的起源
生命之源
微生物是地球上最早的生命形式之一,大约在35亿年前就已经存在。目前普遍认为,微生物是通过自我复制的分 子逐渐演化而来,这一过程发生在地球的原始大气和海洋环境中。
微生物鉴定
通过比较未知微生物与已知微生物的基因序列,可以确定微生物的种类和种群,为疾病 诊断、环境监测等领域提供依据。
微生物生态学研究
生态位分析
微生物群落分析
通过研究微生物在生态系统中的位置和作用, 揭示微生物在生态系统中的功能和相互关系。
通过分析微生物群落的基因序列,了解微生 物群落的组成、结构和动态变化,为环境保 护和生物修复提供指导。
分子系统发育分析是利用分子生物学技 术,通过比较不同微生物的基因序列、 蛋白质序列等分子标记来推断它们的进 化关系。
常用的分子系统发育分析方法包括基因序列 比对、系统发生树构建等,这些方法能够揭 示微生物间的亲缘关系和进化路径。
植物分子系统学研究植物基因组和系统发育的关系
植物分子系统学研究植物基因组和系统发育的关系植物分子系统学是一门研究植物基因组与系统发育关系的学科。
它是通过分析和比较植物的基因组以及基因间的异同,来揭示不同植物之间的亲缘关系和进化路径的科学方法。
本文将探讨植物分子系统学在研究植物基因组和系统发育关系方面的重要性和应用。
植物基因组是植物个体的遗传信息的总和,包含了所有的基因和非编码基因序列。
而系统发育则是研究不同物种之间的进化关系和亲缘关系。
植物分子系统学利用直接或间接的分子数据,如核酸序列,蛋白质序列和基因组结构等,来推断物种之间的演化和亲缘关系。
首先,植物分子系统学能够帮助我们了解植物的进化历史。
通过分析植物基因组的差异和共同点,可以揭示不同植物群体之间的亲缘关系,并推测它们进化的路径。
例如,通过比较不同植物的基因组序列,研究者可以重建植物进化树,从而了解植物界中的不同类群之间的演化关系。
其次,植物分子系统学对植物分类学也有着重要的影响。
传统的植物分类学是基于形态特征的分类方法,但在一些形态上相似的植物中,往往存在基因组差异。
植物分子系统学通过分析植物基因组的特征,能够更准确地确定植物的分类地位和亲缘关系。
这种基于分子数据的分类方法被认为更加客观和准确。
此外,植物分子系统学还对植物的遗传多样性和保护具有重要意义。
研究植物基因组可以帮助我们鉴定和保护濒危物种,通过分析它们的基因组编码,可以了解植物个体之间的遗传差异和亲缘关系,为植物保护和遗传资源的合理利用提供科学依据。
植物分子系统学的研究方法主要包括基因测序技术、分子标记和系统发育分析等。
其中,基因测序技术的广泛应用使得我们能够对植物基因组进行全面测序,并研究不同基因在进化过程中的变化。
分子标记是一种基于植物基因组的遗传标记,通过检测基因组中的可遗传位点,可以进行亲缘关系和遗传多样性的分析。
系统发育分析则是根据不同的分子数据,利用计算机算法和模型,推断植物进化树和亲缘关系。
总的来说,植物分子系统学作为一门跨学科的研究领域,对于我们深入了解植物基因组和系统发育关系具有重要的意义。
序列的同源比较及分子系统学和分子进化分析教学课件
特点
本教学课件内容全面、结构清晰,注重 实践操作和案例分析,有助于学生深入 理解和掌握相关知识。
VS
优势
通过同源比较、分子系统学和分子进化分 析三个方面的内容,使学生对分子生物学 领域有一个全面的了解,同时提高学生的 实验操作能力和解决问题的能力。
感谢您的观看
THANKS
分子系统学与分子进化分析的关联
亲缘关系研究
分子系统学是研究生物亲缘关系和进化顺序的科学,通过比较不同物种或不同基因的分子特征,可以推断出它们 之间的亲缘关系和进化路径。
进化机制研究
分子进化分析是研究生物进化机制的科学,通过比较不同物种或不同基因的分子变异和进化速率,可以揭示生物 进化的内在规律和机制。
它基于氨基酸或核苷酸序列的相似性 比较,以评估物种间的亲缘关系和进 化历程。
序列同源比较的背景
随着生物技术的不断发展,研究人员 能够获得越来越多的基因和蛋白质序 列数据。
为了更好地理解这些数据和物种间的 关系,需要进行序列同源比较,以挖 掘更多有用的信息。
序列同源比较的意义
01
序列同源比较有助于研究物种的进化和亲缘关系。
药物研发
分子系统学研究结果可以用来寻找新的药物靶点,有助于开发出 更加有效的药物。
03
分子进化分析原理
分子进化的概念
分子进化的定义
分子进化是指生物大分子在进化过程中发生的适应性或非适应性 变化的过程。
分子进化的研究内容
主要研究生物大分子演化的规律和机制,包括DNA、蛋白质等分 子的演化过程、速度和方向等。
05
教学课件内容及安排
教学课件的主题和目标
主题
序列的同源比较、分子系统学和分子 进化分析
目标
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
分子系统发育分析用于研究生物体在分子水平的进化式样、方向、速率以及各种分子机制对基因和基因组的结构与功能的影响。
同源——最基本的意义就是具有共同祖先
一般来说,如果两个物种中有两个性状(状态)满足以下两个条件中的任意一个,就可以称这两个性状为一对同源性状:1)它们与这些物种的祖先类群中所发现的某个性状相同;(2)它们是具有祖先—后裔关系的不同性状。
同源性一般是指核酸分子的核苷酸序列之间或蛋白质分子的氨基酸序列之间的相似程度。
直系同源(rothology)可反映五种血统上的同源性,既物种进化的历史。
祖先类群:如果一个类群或物种至少有一个子裔类群,这个原始类群就是祖先类群。
单系类群:包含一个祖先类群所有子裔的群组称为单系类群。
并系类群:不满足单系类群要求,各成员间又具有共通祖先特征的群组。
姊妹群:与某一类群在谱系关系上最为密切的类群称为姊妹群。
内类群和外类群:一项研究所涉及的某一特定类群可称为内类群,不包括在内类群中又与之有一定关系的类群可称为外类群。
序列分析是最终测定同源性程度的方法。
DNA-DNA杂交或DNA-RNA杂交也是有用的估计途径。
在分子系统发育分析中,首先应考虑直系同源基因序列。
系统树(phylogenetic tree) :用来表达类群(或序列)间系统发育关系的一种树状图。
可划分为以下几种类型: 有根树(rooted tree)和无根树(unrooted tree) 以外类群作为树根的系统树称为有根树;没有外类群为树根的系统树称为无根树。
有根树数目的计算方法:Nr=(2n—3)!{2n-2(n—2)!}
无根树数目的计算方法:Nu=(2n—5)!{2n-3(n—3)!}
基因树(gene tree)是由一个基因所构建的系统树。
物种树(species tree):则表达了某一特定类群的进化路径。
核苷酸置换模型可以用4X4的矩阵表示。
估算两个蛋白质序列间置换数的方法中必须将同义置换和非同义置换非分开考虑,而起始和终止密码子应排除在外因为它们几乎不随时间变化。
判断是同义置换还是非同义置换,关键是看翻译结果。
即置换后翻译的氨基酸是否有变。
核苷酸序列分歧度:DNA序列间的分歧度k是一种相异性指数,可通过序列成对比较获得碱基差异值,然后运用序列进化模型来校正。
蛋白质编码序列分歧度:设Ks为两个序列间同义变化的分歧度,KA为非同义变化的分歧度,应用但参数模型。
可以计算。
系统树构建方法简约法,相容法,距离距阵法(包括邻接法和UPGMA法),最大似然法。
UPGMA法:使用算术平均的不加权对群法的缩写,也称为平均法,是目前聚类分析中使用的最多的一种聚合策略。
统计检验工具有重复取样法(分为自展法和刀切法)。