大学化学第四章计算
大学有机化学课件第四章 芳香烃

定条件下还是能加成的。
加氢:
加氯: 3.氧化反应 苯环侧链的氧化: 烃基苯侧链可被高锰酸钾或重铬酸钾的酸性或碱性溶液或稀硝酸所氧化,并在与苯环直接相连 的碳氢键开始,如果与苯环直接相连的碳上没有氢时,不被氧化。氧化时,不论烷基的长短,最后 都变为羧基,苯环不容易氧化。
苯环的氧化:
第四节 苯环的亲电取代定位规律 一、定位规律 1. 定位基在苯环上引入新的取代基时,其进入苯环的位置,主要决定于原有取代基的性质。这个原有 的取代基称为定位基。 2. 定位基类型 第一类:邻、对位定位基,使反应容易进行,并使新导入基主要进入苯环的邻、对位。 如:-NH2、-ph、- N(CH3)2 、-NHCOCH3 、-OH、- OCH3 第二类:间位定位基,使反应难于进行,并使新导入基进入苯环的间位。 + 如:-COOH 、-NO2 、- N(CH3)3 、-CF3 第三类:使反应较难进行,又使新基导入邻位或对位。 如:-F、- Cl、- Br 、-CH2Cl 二、定位规律的解释 1. -COOH 、-NO2 、- N(CH3)3 、-CF3等。 这类定位基与苯环直接连接的原子都具有一定的正电荷,吸引苯环上的电子,使苯环上的电子 云降低,使亲电取代反应较难进行。以硝基为例,硝基的π轨道和苯环构成π-π共轭体系,由于 氧、氮的电负性强于碳,使共轭体系的电子云移向硝基。诱导效应和共轭效应协同作用的结果,降 低了苯环的电子云密度,其中以邻、对位为甚,而间位相对来说降低的少一些。
+ 3H 2
二、近代物理方法测定苯的结构
+Q
应该 Q=120×3=360KJ/mol,而实际上苯的氢化热=208 KJ / mol,比理论值低 152 KJ / mol 。
1.近代物理方法测定苯的结构: (1)6 个碳组成一个平面正六边形,6 个氢与 6 个 碳都在同一个平面上。 (共平面性) (2)所有键角都是 120° (3)碳碳键键长为 0. 139nm. 2.轨道杂化理论: 苯环所有碳原子都是采用 SP2 杂化,每个碳原子以三个杂化轨道分别与相邻的碳原子和氢原子形成 三个σ键。每个碳原子的未参与杂化的 P 轨道都垂直于碳环的平面。相邻的两个 P 轨道彼此从侧面 重叠,形成一个封闭的共轭体系,这个封闭的共轭体称为大π键,由于π电子高度离域,从而使键 达到完全平均化。
大学化学 化学平衡

多重平衡规则。
如果 反应(3) = 反应(1) + 反应(2)
Ө Ө K3 = K1 · 2Ө K
如果 反应(4) = 反应(1) – 反应(2)
Ө K4 = K1 /K2 Ө
Ө
4.1. 书写平衡常数表达式的规则
[例题1] 已知在298K时:
(1) H2(g) + S(s) (2) S(s) + O2(g) 求反应H2(g) + SO2(g) KӨ=? 解:反应(1) – (2) H2(g) + S(s) –S(s) –O2(g) H2(g) + SO2(g) H2S(g) –SO2(g) H2S (g) SO2 (g) K1 =1.0×10-3
[例题1]
计算上述反应1号和2号实验中碘的转化率分别 是多少?
5. 平衡常数与转化率
1号实验中碘的转化率为:
碘的转化率
( 1 1 .9 6 3 .1 2 9 ) m o l L 1 1 .9 6 m o l L
-1 -1
100%
8 .8 3 1 1 1 .9 6
1 0 0 % 7 3 .8 %
[HI]2 [H2]· 2] [I
1 2
3 4
10.67 11.34
0 0
11.96 7.510
0 0
0
1.831 4.565
0.4798 1.141
3.129 0.7378
0.4798 1.141
17.67 13.54
3.531 8.410
54.50 54.43
54.16 54.33
0
4.489 10.69
南京大学物理化学(第五版)04章_多组分系统热力学
(
Gm p
)T
Vm
对多组分系统,把 Gm 换为 B ,则摩尔体积变为偏
摩尔体积 VB 。
化学势与温度的关系
(
B
T
)
p
,nB
,
nc
[ T
G ( ) ] T , p,nc p,nB ,nc nB
[ nB
G ( T ) p,nB ,nc ]T , p,nc
(S) [ nB ]T , p,nc
nk 0
dnk
k
n1Z1 n2 Z2 nk Zk nBZB B=1
偏摩尔量的加和公式
k
Z= nB ZB
B=1
这就是偏摩尔量的加和公式,说明系统的总 的容量性质等于各组分偏摩尔量的加和。
例如:系统只有两个组分,其物质的量和偏 摩尔体积分别为 n1,V1 和 n2 ,V2 ,则系统的总体积为:
(1)热力学能
设系统中有 1, 2,3, , k 个组分
所含的量分别为 n1, n2, , nk
U U (S,V , n1, n2, , nk )
化学势的定义
U U (S,V , n1, n2, , nk )
其全微分为
dU
U ( S )V ,nB dS
(
U V
)
S
,nB
dV
k U B1 ( nB )S ,V ,nc(cB) dnB
如果转移是在平衡条件下进行,则
dG 0 又
dnB dnB
所以 (B B )dnB 0
化学势在相平衡中的应用
(B B )dnB 0
因为 dnB 0 所以
B B
组分B在α,β两相中,达平衡的条件是该
第四章共轭结构和二烯烃20150908-合肥工业大学-有机化学

活化能高,但生成结构稳定的产物。--高温反应
2020/12/19
10
3.2 双烯合成反应 (1)定义
固体
顺-1,2,5,6-四氢化邻苯二甲酸酐(100%) 鉴别共轭二烯烃
在光或热的作用下,共轭二烯烃与含有吸电子基团(-CHO、 -COR、-CO2R、-CN、NO2等)的双键或叁键的化合物进行1,4-加成反应,生成环状化合物。周环反应的一种。
域选择性反应。
2020/12/19
13
立体选择性:立体专一、顺式加成
H CO OH +
HO OC H
COOH
H H
+
COOH
H
CO OH CO OH H
立体选择型反应指的是当一个有机反应可能产生几个立体异构体时,其中一个或多个的 立体异构体优先获得的反应。
2020/12/19
14
双烯合成反应举例:
B r
C H 2C HC HC H 2
1,2-加成
B r
C H 3C HC HC H 2
H 2 C 1 C 2HC 3HC 4H 2+H B r
1,4-加成
B r C H 2C HC HC H 3
1,2-加成: 亲电试剂(溴)加到共轭烯烃其中一个双键上,即C-1和C-2上。
1,4-加成: 亲电试剂(溴)加到C-1和C-4上(即共轭体系的两端),双键移到中间,称1,4-加成或共
还有一种特殊的情况是由于组成分子轨道的原子轨道的空间对称性不匹配原子轨道没有有效重叠组合得到的分子轨道的能量跟组合前的原子轨道能量没有明显差别所得的分子轨道叫做非键分子轨道
章二烯烃和有机分子中电荷效应
节二烯烃结构和性质
1、二烯烃的分类和命名 二烯烃:分子中含有两个C=C双键的烃。 通式:CnH2n-2n≥3
大学基础课程有机化学课件第四章二烯烃

CH2
Br H CHCHCH2
E1,4
E1,2
C H 3 C HC H 3C H 3
Br
H
活化能:
H2C CHCH CH3
B r C H 2 C H C H C H 3
Br
E1,4 > E1,2
稳定性:
图4.9
反应进程
1,2–加成与1,4–加成势能图
产物1,2 <
产物1,4
1,2–加成产物:反应速率控制或动力学控制; 1,4–加成产物:反应温度控制或热力学控制.
4.3.2 p,π–共轭
烯丙基正离子(allylic carbocation):
C H 2 C HC H 2
+
图 4.5 烯丙基正离子 的p,π–共轭
带有正电荷的C原子: sp2杂化,空的p 轨道 与π轨道在侧面进行 相互交盖,电子发生 离域.
碳正离子的稳定性: C H 3
C H 3C HC HC H C H 3>C H 3 C>H 2CC HC H 2
s–反式 构象
s-cis-conformation s-trans-conformation
4.3 电子离域与共轭体系
电子离域:1,3-丁二烯分子中,四个π电 子不是两两分别固定在两个双键碳原子之 间,而是扩展到四个碳原子之间,这种现象 称为电子的离域.〔电子的离域体现了分子 内原子之间相互影响的电子效应〕 这样的分子称为共轭分子. 这样的体系称为共轭体系.
C原子之间,而是分布在共轭
体系中的几个C原子上.
键长趋于平均化
降低了分子的能量,提高了体系的稳定性
二烯烃
氢化热/(kJ·mol-1)
C H 3C H C H C H C H 2 226 1,3–戊二烯
大学有机化学第四章 环烃

目前有关苯的结构还没有更好的表达方式, 一般沿用的有: 或
物理性质 (physical property) 多数是无色有芳香味的液体,易挥发,蒸汽有毒(也 有的芳香烃很臭!)。相对密度小于1,不溶于水,能溶 于乙醇、乙醚、丙酮等有机溶剂。 化学性质 (chemical property) 芳香烃具有其特征性质——芳香性(易取代,难加 成,难氧化)。 1.取代反应 (1)卤代
3. 二元取代物的定位效应 1)原有取代基定位效应一致,第三个取代基进入它们 共同确定的位置。
CH3 (>99%)
COOH
COOH Br
NO2
(100%)
SO3H
(80%)
(20%)
2)两个取代基定位效应相矛盾,由基团致活能力顺序 判断第三个基团取代的位置。
①.两个基团不同类,定位效应受邻对位取代基控制。
HONO2 + HOSO2OH
[H2O+-NO2] + SO4H - H2 O NO2
H
- H+
+ NO2 π络合物 H+ + SO4H
NO2 σ络合物
NO2
NO2
H2SO4
3.磺化
2 H2SO4
O
SO3
S
O
δ
+ H3O+ + HSO4
H
+
δ
H+
SO3H
O
SO3
苯环上取代基的定位规律(定位效应或称取向效应)
X X X
缺点:3.
一般单、双键的键长分别为 : 0.154nm 和0 .134 nm而实际苯分子是一个正六边 形构型,且碳碳间键长完全相等,均为 0 .139nm (介于一般单、双键键长之间)
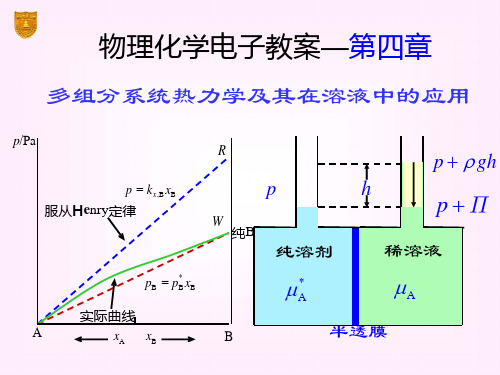
南京大学物理化学 第四章 多组分均相系统热力学及其在溶液中的应用
大能力(可逆时系统对外所做功最大) ② 当W’=0时,:反应永远向着化学势降低的方向进行,可用来判断
反应进行的情况(=0可逆,<0不可逆) 该判据也可推广到多组分多相系统:和 2. 判据的应用 (1) 相变
广义的相变是物质由一个相迁往另一个相的过程,是一个物质流动的 过程。
第15次课
3. 理想溶液的化学势
化学势是物质迁移的推动力,不论物质是否混合,只要气液两相平 衡,则气液两相的化学势相等。 混合前:纯组分 混合后:溶液组分
是纯液体A在温度为T、压力为溶液上方总压时的化学势。
4. 理想溶液的热力学性质 (1) 蒸气压与液相组成的关系
,故 (2) 蒸气压与气相组成的关系
等温等压条件下,非挥发性溶质形成的溶液中,溶剂的蒸气压等于 纯溶剂的蒸汽压乘以溶液中溶剂的摩尔分数(或:溶剂蒸气压的降低值 与纯溶剂的蒸气压之比等于溶质的摩尔分数) 说明:公式只适用于溶液中只有A、B两个组分的系统(),而则具有 普适性。
2. 亨利定律(Henry) 一定温度、压力下,稀溶液中某挥发性物质的平衡分压与该溶质的
① ∵∴压力p升高,化学势μB也随之增加 ② 若已知,则可求出 说明:实际上,在与吉布斯自由能有关的关系式中,如果把G换成μ, 并将公式中其它广度量换成相应的偏摩尔量,则公式仍然成立。 例:
第14次课
(四)化学势判据
1. 判据推导 组成可变的封闭系统,发生广义化学变化时(可逆取等号) 将上述四式与⑤-⑧式对比,得到
可称为定浓物理量
2 偏摩尔量的集合公式(加和定理) 等温等压条件下,
在任一系统中,将各组分的物质的量增加一倍,其各组分浓度仍不 变,广度量Z则相应增加一倍。 注:在所有偏摩尔量中,只有偏摩尔体积可测,可由求出溶液的总体 积。
大学普通化学课件
酸
HAc
H
2
PO
4
HPO
24
NH
+ 4
[Al(H 2O) 6] 3+
[Al(OH)(H 2O) 5 ]2+
H+ +碱
H + + Ac -
H+
+
HPO
24
H+
+
PO
34
H + + NH 3
H + + [Al(OH)(H 2O) 5]2+
KW =1.0×10-14
100℃纯水:KW =5.43×10-13 T , KW
4.2.2 溶液的pH
pH = -lg{c(H3O+ )} 令 pOH = -lg{c(OH- )}
常温下,
根据
KW
={c(H
O+
3
)}{c(OH
-
)}=
1.0×10-14
即 - lg c(H+ ) - lg c(OH- ) = - lg KW = 14
H3O+(aq) +OH-(aq) 酸(2) 碱(1)
③ 盐类水解反应也是离子酸碱的质子 转移反应。例如NaAc水解:
H+
Ac-+H2O OH- + HAc 碱(1) 酸(2) 碱(2) 酸(1)
NH4Cl水解:
H+
NH
+ 4
+
H2O
H3O+ + NH3
酸(1) 碱(2) 酸(2) 碱(1)
山东大学期末考试复习 水分析化学[第四章沉淀滴定法]山东大学期末考试知识点复习
第四章沉淀滴定法一、沉淀滴定法是以沉淀反应为基础的滴定分析法通常应用最多的是银量法,银量法主要用于测定水中C1一、Br一、I一、Ag+及SCN一等。
因所用指示剂不同可分为莫尔法(Mohr)、佛尔哈德法(Volhard)、法扬司法(Fajans)三种。
莫尔法:以铬酸钾作为指示剂的银量法称为莫尔法。
本法适用于测定Cl一、Br一和Ag+。
一般控制溶液为中性或弱碱性。
佛尔哈德法:是用铁铵钒即硫酸高铁铵作为指示剂的银量法。
有直接滴定法和返滴定法。
以NH4SCN或KSCN为滴定剂。
佛尔哈德法可用于测定C1一、Br、I 一、Ag+及SCN一。
佛尔哈德法最大的优点是可在酸性溶液中进行滴定,且方法选择性高。
但测定卤素离子时需使用AgNO3和NH4SCN两种标准溶液。
在测定Cl一时,需加入有机溶剂以防止沉淀发生转化反应。
法扬司法:是用吸附指示剂指示滴定终点的银量法。
吸附指示剂是一类有机染料(如荧光黄),在溶液中可离解为具有一定颜色的阴离子,此阴离子容易被带正电荷的胶体沉淀所吸附,从而引起颜色的改变,指示终点到达。
法扬司法可测定Cl、Br一、I一、Ag+及SCN一,一般控制溶液为弱酸性或弱碱性。
法扬司法方法简便,终点明显,但反应条件要求比较严格,应注意溶液的酸度、浓度及胶体的保护等。
二、重量分析法概述1.重量分析法,一般将被测组分与试样中的其他组分分离,转化为一定的称量形式,称量后,计算得出被测组分的含量。
根据被测组分与其他试样分离方法的不同,重量法可分为:沉淀法、气化法、电解法和萃取法。
本章主要介绍沉淀法。
沉淀法是利用沉淀反应使被测组分以沉淀形式析出,通过过滤、洗涤、烘干或灼烧后,称量并计算被测组分的含量。
2.重量分析对沉淀形式和称量形式的要求。
利用沉淀反应,使被测组分以适当的“沉淀形式”析出,过滤、洗涤后再将沉淀烘干或灼烧成为“称量形式”称量。
沉淀形式和称量形式可能相同,也可能不相同。
重量分析对沉淀形式的要求:①沉淀的溶解度要足够小;②沉淀的纯度高;③沉淀易于洗涤和过滤;④沉淀易于转化为具有固定组成的称量形式。
大学化学:练习册习题及答案第四章
第四章电化学一.判断题(正确的画“∨”,错误的画“×”)1.在相同条件下,氧化还原电对中电极电势代数值愈小的还原态,其还原能力愈强。
2.在氧化还原反应中,凡是ϕθ值小的氧化态一定不能氧化ϕθ值大的还原态。
3.ϕ值仅与物质的本性有关。
4.在298K下,ϕθ值与物质的本性有关。
5.一定温度下,在氧化还原电对中氧化态的浓度降低,则还原态的还原能力增强。
6.一定温度下,在氧化还原电对中还原态的浓度增加,则氧化态的氧化能力减弱。
7.巳知半反应H2O2→O2+2H++2e-,过氧化氢是该半反应中的氧化态物质。
8.对于电极反应I2+2e-→2I-其ϕθ=0.54V,将反应改写为1/2I2+e-→I-,则ϕθ=0.27V。
9.微小浓度的改变就很容易逆转的氧化还原反应,是那些Eθ值接近于零的反应。
10.当一种氧化剂能氧化系统中的几种还原剂时,首先发生的反应一定是在E值大的电对之间。
11.已知电对Br2/Br-,Fe3+/Fe2+,I2/I-的ϕθ值分别为1.065V、0.771V、0.535V,则它们中氧化态氧化能力的顺序是:Br2>Fe3+>I212.一定温度下,Cr2O2-7的氧化性随溶液的pH值增大而增强。
13.氧化还原反应Cu+2Ag+=Cu2++2Ag,改写为1/2Cu+Ag+=1/2Cu2++Ag在标准状态下Eθ不变。
14.将13题的方程式改写为:Cu2++2Ag=Cu+2Ag+在标准状态下Eθ不变。
15.在标准条件下,反应:2MnO4-+10 Cl-+16H+=2 Mn2++5 Cl2+8H2O的原电池图式为:(-)Pt|Cl2|Cl-‖MnO4-,Mn2+,H+(cθ)|Pt(+)16.巳知ϕθ(Fe3+/Fe2+)=0.77V,ϕθ(Sn4+/Sn2+)=0.15V,则氧化还原反应进行的方向为:Sn4++Fe2+ = Sn2++2 Fe3+(在标准条件下)17.锌的浓差电池,其原电池符号为:(-)Zn|Zn2+(1.0 mol·L-1) ‖Zn2+(0.0010 mol·L-1) ∣Zn(+)18.巳知ϕθ(Br2/Br-)=1.07V,ϕθ(Fe3+/Fe2+)=0.77V,在标准条件下则Br-的还原能力较Fe2+强。