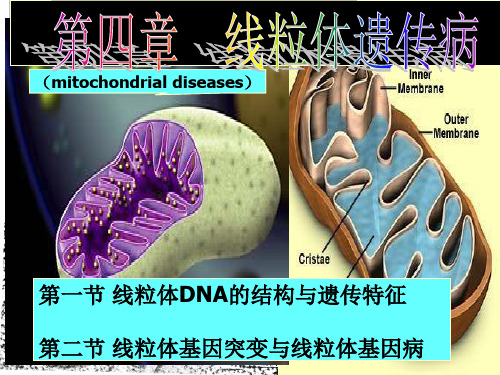
线粒体遗传病
线粒体的遗传规律和发病规律

线粒体的遗传规律和发病规律一、引言线粒体是细胞内的一种特殊细胞器,其主要功能是参与能量代谢过程。
线粒体拥有自己的基因组,称为线粒体DNA(mtDNA),其遗传规律与核基因不同。
本文将介绍线粒体的遗传规律和发病规律。
二、线粒体的遗传规律1. mtDNA的结构mtDNA是环状分子,大小约为16.6kb,含有37个基因,其中13个编码蛋白质、22个编码tRNA和2个编码rRNA。
mtDNA存在于线粒体内,每个线粒体通常含有2-10份mtDNA分子。
2. mtDNA的复制方式mtDNA复制是由多种酶和蛋白质协同完成的。
在细胞分裂时,每个新细胞都会获得一定数量的线粒体,并且这些新细胞中每一个线粒体都会含有相同数量和类型的mtDNA分子。
3. mtDNA的遗传方式mtDNA是通过母亲遗传给下一代。
这是因为在受精卵形成过程中,只有母亲提供了大部分线粒体和其中的mtDNA。
父亲提供的只有少量或没有线粒体和mtDNA。
4. mtDNA突变由于mtDNA的复制方式不同于核基因,因此其突变率也比较高。
这些突变可能会导致线粒体功能障碍和疾病的发生。
三、线粒体的发病规律1. 线粒体疾病的分类线粒体疾病是一类由mtDNA遗传突变引起的遗传性疾病,主要包括以下几种类型:(1)线粒体脑肌(MELAS)综合征:主要表现为中枢神经系统和肌肉功能障碍。
(2)线粒体脑卒中样发作(MERRF)综合征:主要表现为抽搐、共济失调、视力障碍等。
(3)非典型家族性震颤麻痹(NARP):主要表现为神经系统和肌肉功能障碍。
2. 线粒体疾病的诊断方法目前,诊断线粒体疾病主要依靠以下几种方法:(1)临床表现:根据患者的临床表现进行初步判断。
(2)遗传学检测:通过对患者和家族成员的mtDNA进行分析,确定是否存在突变。
(3)生化检测:通过对血液、尿液等样本的生化指标进行分析,确定是否存在线粒体功能障碍。
3. 线粒体疾病的治疗方法目前,线粒体疾病的治疗方法主要包括以下几种:(1)对症治疗:根据患者的临床表现进行对症治疗。
有关线粒体病的PPT大纲

与其他累及脑和肌肉的疾病进行鉴别,如多发性硬化、重症肌无力等。同时,注 意与线粒体病其他类型进行鉴别,如线粒体肌病、线粒体神经病等。
治疗策略及预后评估因素
治疗策略
目前尚无特效治疗方法,主要采用对症治疗和支持治疗。如给予抗癫痫药物控制癫痫发作,给予营养支持改善营 养不良等。同时,基因治疗和细胞治疗等新型治疗方法正在研究中。
临床表现与分型
慢性进行性眼外肌麻痹(CPEO)
主要表现为双眼眼睑下垂、眼外肌麻痹、眼球运动障碍等。
Kearns-Sayre综合征(KSS)
表现为眼外肌麻痹、视网膜色素变性、心脏传导阻滞和脑脊液蛋白升 高等。
MELAS综合征
即线粒体脑肌病伴乳酸血症和卒中样发作,表现为肌无力、癫痫、智 力低下、卒中样发作等。
线粒体脑肌病常累及中枢神经系统,导致神经元能量代谢障碍,引发一系列神经系统症状 。
临床表现与分型特点
MELAS综合征 MERRF综合征
KSS综合征 Leigh综合征
以线粒体脑病、乳酸酸中毒和卒中样发作为主要特点,患者可 出现偏瘫、失语、认知障碍等。
以肌阵挛性癫痫、小脑共济失调和智能障碍为主要表现,患者 可出现肌肉抽搐、站立不稳、智力下降等。
预防措施建议
遗传咨询
对于有家族史的高危人群,应进 行遗传咨询和基因检测,以明确 遗传风险和制定预防措施。
避免近亲结婚
近亲结婚可能增加遗传病的发生 风险,因此应避免近亲结婚以降 低线粒体病的发病率。
孕期保健
孕妇在孕期应保持良好的生活习 惯和饮食营养,避免接触有害物 质和环境,以降低胎儿患线粒体 病的风险。
06
患者管理与康复支
持
患者心理干预与家庭支持
心理干预策略
线粒体病名词解释

线粒体病名词解释线粒体病是一类常见的遗传病,它们是由线粒体遗传物质或基因突变引起的。
线粒体是一种细胞内细胞器,它负责细胞内同化和分解物质,以提供能量。
这些病症可能影响不同年龄段的人,并且可以大多数患者都损害致命。
一般来说,线粒体病是一类常见的遗传疾病,可以归类为非常少数的家族遗传疾病,可能会给患者的生活带来极大的影响,从而导致可能的残疾。
线粒体病的症状也可能根据疾病的种类而有很大不同,并且疾病的治疗方式也会因病症的不同而有所不同。
线粒体衰老现象 (mitochondrial aging), 也称为mitochondrial dysfunction,是指线粒体功能下降所引起的一种症状。
线粒体衰老可能导致一系列症状,如疲劳、嗜睡、认知障碍、疼痛和神经系统紊乱等。
此外,线粒体病也可以引发多系统破坏,如心肌病、眼病、肝病和消化系统疾病等。
线粒体病还可能与一些其他疾病有关,如糖尿病、神经退行性疾病和特发性肝病等,这些疾病在一定程度上都受到线粒体紊乱的影响。
因此,建立正确的线粒体健康概念,制定有效的预防和治疗策略,对改善患者的病情和生活质量具有重要意义。
线粒体病一般可以根据症状和遗传背景进行诊断,例如病史和家族史,但检查也很重要。
一般来说,会进行一些检查,如血液检查、肌酐检查等,这些检查可以帮助医生们更好地诊断疾病,而基因检查和线粒体检查也可以检测出病因。
线粒体病的治疗可以根据疾病的特点而有所不同,但一般来说,患者会接受药物治疗、营养支持和康复治疗。
除此之外,患者也可以接受基因治疗或细胞治疗,以及机器人技术和外科技术等其他治疗方法。
此外,线粒体病的治疗还包括一些其他因素,如心理支持和社会支持等,这些可以让患者拥有更好的生活质量,以及更多的机会去改善病情。
总的来说,线粒体病是一类常见的遗传病,它的症状和疾病的治疗方式也会因病症的不同而有所不同。
因此,对于线粒体病的检测、治疗和预防,需要进行更有效的干预,以改善患者的病情和提高他们的生活质量。
第四章线粒体遗传病课件
•线粒体的代谢障碍,则不能产生足够的能量而导 致细胞功能衰退,出现一系列临床症候。人群患病 率约为1/8,500
线粒体基因突变 表现的临床特征:
线粒体突变导致的疾病主要 累及中枢和外周神经系统, 肌病和脑病症状。与贫血和
糖尿病等疾病也相关。
问题:线粒体疾病主要受累的器官是哪些
•(一)碱基突变
•1、错义突变:称氨基酸替换突 变。 Leber遗传性视神经萎缩.
• 线粒体病有累加效应因此线粒体病 有随着年龄的增加病情会越来越严重 的特征。
• 问题:什么叫阈值效应?
(六)mtDNA的突变率极高
mtDNA的突变率比核DNA高10~20倍。 但因为都是中性和中度有害的mtDNA的 突变,有害的突变会通过选择(例如遗传 瓶颈) 而消除,故线粒体遗传病并不常 见。
相关,如线粒体肌病和脑肌病、线粒体眼病, 老年性痴呆、帕金森病、Ⅱ型糖尿病、心肌 病及衰老等,有人统称为线粒体疾病。
•
复习思考题:
• 1、线粒体DNA的结构特征如何?
• 2、简述线粒体遗传的特点。
• 3、线粒体疾病主要受累的器官是哪些?
• 4、常见的线粒体遗传病有哪几种?它们有什么共同特征?
• 5、何谓多基因遗传方式?
•2、蛋白质生物合成基因突变: 为tRNA基因突变,
(二)缺失、插入突变: 例如,肌阵挛性癫痫
(MERRF综合征)等
•例如,眼肌病,如KSS综 合征(无家族史、散发)
mtDNA拷贝数目低 于正常。例如致死 (三)mtDNA拷贝数目突变:性婴儿呼吸障碍等
突变 mt-3243 mt-3256 mt-3271 mt-3303 mt-3260 nmt-4269 mt-5730 nmt-8344 mt-8356 mt-15990 mt-8993 mt-11778 mt-4160 mt-3460 mt-7444 mt-14484 mt-15257
线粒体遗传与线粒体疾病

第八章线粒体疾病的遗传线粒体是真核细胞的能量代谢中心,其内膜上富含呼吸链-氧化磷酸化系统的酶复合体,可通过电子传递和氧化磷酸化生成A TP,为细胞提供进行各种生命活动所需要的能量。
大量研究表明,线粒体内含有DNA和转译系统,能够独立进行复制、转录和翻译,是许多人类疾病的重要病因。
第一节人类线粒体基因组线粒体DNA(mitochondrial DNA,mtDNA)是独立于细胞核染色体外的又一基因组,被称为人类第25号染色体,遗传特点表现为非孟德尔遗传方式,又称核外遗传。
mtDNA分子量小,结构简单,进化速度快,无组织特异性,具有特殊的结构特征、遗传特征和重要功能,而且在细胞中含量丰富(几乎每个人体细胞中都含有数以百计的线粒体,一个线粒体内有2~10个拷贝的DNA),易于纯化,是研究基因结构和表达、调控的良好模型,在人类学、发育生物学、分子生物学、临床医学、法医学等领域受到广泛的重视,并取得令人瞩目的成就。
1981年,Anderson等人完成了人类线粒体基因组的全部核苷酸序列的测定。
mtDNA所含信息量小,在呼吸链-氧化磷酸化系统的80多种蛋白质亚基中,mtDNA仅编码13种,绝大部分蛋白质亚基和其他维持线粒体结构和功能的蛋白质都依赖于核DNA(nuclear DNA,nDNA)编码,在细胞质中合成后,经特定转运方式进入线粒体。
此外,mtDNA基因的表达受nDNA的制约,线粒体氧化磷酸化酶系统的组装和维护需要nDNA和mtDNA的协调,二者共同作用参与机体代谢调节。
因此线粒体是一种半自主细胞器,受线粒体基因组和核基因组两套遗传系统共同控制(图8-1),nDNA与mtDNA基因突变均可导致线粒体中蛋白质合成受阻,细胞能量代谢缺陷。
一、线粒体基因组的结构线粒体基因组全长16569bp,不与组蛋白结合,呈裸露闭环双链状,根据其转录产物在CsCl中密度的不同分为重链和轻链,重链(H 链)富含鸟嘌呤,轻链(L链)富含胞嘧啶。
线粒体遗传病

线粒体疾病的遗传一、线粒体的功能:✧是细胞有氧呼吸的基地和供能的场所,供应细胞生命活动95%的能量✧线粒体的主要功能是把氧化各种底物产生的自由能转化为可被细胞直接利用的形式——ATP✧细胞氧化(细胞呼吸)✧无氧酵解:1分子葡萄糖→2ATP 线粒体有氧呼吸:1分子葡萄糖→36~38ATP二、mtDNA的遗传特点:1、具有复制半自主性。
(M染色体,25号染色体)线粒体内含有DNA分子,被称为人类第25号染色体,是细胞核以外含有遗传信息和表达系统的细胞器,其遗传特点表现为非孟德尔遗传方式,又称核外遗传。
2、部分遗传密码与核DNA不同。
3、母系遗传。
(不符合经典遗传定律)。
精卵结合时,受精卵中的线粒体DNA几乎全都来自于卵子,来源于精子的mtDNA 对表型无明显作用,这种双亲信息的不等量表现决定了线粒体遗传病的传递方式不符合孟德尔遗传,而是表现为母系遗传(maternal inheritance),即母亲将mtDNA传递给她的儿子和女儿,但只有女儿能将其mtDNA传递给下一代。
4、在细胞分裂间期经过复制和分离。
细胞分裂时,突变型和野生型mtDNA发生分离,随机地分配到子细胞中,使子细胞拥有不同比例的突变型mtDNA分子。
5、具有阈值效应。
在克隆和测序的研究中发现一些个体同时存在两种或两种以上类型的mtDNA,这是由于mtDNA发生突变,导致一个细胞内同时存在野生型mtDNA和突变型mtDNA,称为“杂质”(heteroplasmy)。
野生型mtDNA对突变型mtDNA有保护和补偿作用,因此,mtDNA突变时并不立即产生严重后果。
突变所产生的效应取决于该细胞中野生型和突变型线粒体DNA的比例,只有突变型DNA达到一定数量(阈值)才足以引起细胞的功能障碍,这种现象称为阈值效应。
阈值效应的一个表现就是在某些线粒体遗传病的家系中,有些个体起初并没有临床症状,但随年龄增加由于自发突变、环境选择等原因,突变型DNA逐渐积累,线粒体的能量代谢功能持续性下降,最终出现临床症状。
线粒体的遗传规律和发病规律
线粒体的遗传规律和发病规律引言线粒体是细胞内的一个重要细胞器,它参与能量代谢和细胞生命活动的调控。
线粒体不仅拥有自己的DNA(线粒体DNA,mtDNA),还具有一定的遗传规律。
研究发现,线粒体的遗传规律与发病规律密切相关,对于相关疾病的治疗和预防具有重要意义。
线粒体的遗传规律线粒体的遗传规律主要涉及到线粒体DNA的传递和突变。
1. 线粒体DNA的传递线粒体DNA的传递方式与细胞核DNA有所不同。
在人类细胞中,细胞核DNA是由父母双方各贡献一半的基因;而线粒体DNA只由母亲传递给后代。
这是因为在受精卵中,细胞质中的线粒体是由卵细胞提供的,而精子贡献的细胞质被几乎完全丧失。
因此,线粒体的遗传是单亲遗传,只由母系传递给子代。
2. 线粒体DNA的突变线粒体DNA的突变是常见的,突变率比细胞核DNA高。
线粒体DNA的突变主要是由于其自身复制的特点所致。
与细胞核DNA不同,线粒体DNA的复制缺少校对修复机制,容易产生突变。
此外,线粒体DNA还很容易受到氧化应激和其他损伤因素的影响,导致进一步的突变。
线粒体的发病规律线粒体的突变与多种疾病的发生发展密切相关。
主要包括线粒体遗传病和一些与线粒体功能紊乱相关的疾病。
1. 线粒体遗传病线粒体遗传病是由于线粒体DNA突变导致的遗传性疾病。
常见的线粒体遗传病包括Leber遗传性视神经病变(LHON)、MERRF综合征、MELAS综合征等。
这些疾病的发生与线粒体功能异常有关,影响了线粒体能量代谢的过程,导致器官和组织的损伤。
线粒体遗传病表现出明显的家族性遗传特点,且发病年龄和症状表现不一致。
2. 与线粒体功能紊乱相关的疾病除了线粒体遗传病外,线粒体功能紊乱还与一些其他疾病的发生相关。
例如,心肌梗死、中风、癌症等疾病的发生与线粒体功能异常有一定关联。
线粒体功能异常导致能量代谢的紊乱,进而影响细胞的正常生理功能,最终导致各种疾病的发生。
线粒体疾病的诊断和治疗线粒体疾病的诊断和治疗对于患者的生存和生活质量具有重要意义。
第06章-线粒体遗传病
Complex Ⅰ Ⅱ Ⅲ Ⅳ
Subunits 41 4 11 13
Nuclear 34 4 10 10
mtDNA 7 0 1 3
ATPase
14
12
2
合计
87
70
13
(二)线粒体基因组所用的遗传密码
和通用密码不完全等同
Codon UGG UGA AGG AGA AUG AUA
Universal code Trp Stop Arg Arg Met Ile
含16569个碱基对。 外环为重链(H)富含G
12种多肽链 12S rRNA 16S rRNA 14种tRNA
内环为轻链(L)富含C
1种多肽链 8种tRNA
mtDNA共有37个 基2因种 编码
rRNA(12S和16S )基因
22种 编码 tRNA 基因
13种 编码 蛋白质 基因
Human mtDNA, a circular molecule that has been completely sequenced, is among the smallest known mtDNAs, containing 16,569 base pairs. It encodes the two rRNAs found in mitochondrial ribosomes and the 22 tRNAs used to translate mitochondrial mRNAs.
(一)线粒体蛋白输入缺陷 (二)底物运输缺陷 (三)底物利用缺陷 (四)铁运输缺陷 (五)电子传递链缺陷
mtDNA Trp Trp Stop Stop Met Met
(三)mtDNA为母系遗传
母亲将她的mtDNA 传递给儿子和女儿, 但只有女儿能将其 mtDNA传递给下一代。
医学遗传学习题(附答案)第6章 线粒体遗传病
第六章线粒体遗传病(一)选择题(A型选择题)1.下面关于线粒体的正确描述是______。
A.含有遗传信息和转译系统B.线粒体基因突变与人类疾病基本无关C.是一种完全独立自主的细胞器D。
只有极少量DNA,作用很少E.线粒体中所需蛋白质均来自细胞质2. 关于线粒体遗传的叙述,不正确的是______。
A.线粒体遗传同样是由DNA控制的遗传B.线粒体遗传的子代性状受母亲影响C.线粒体遗传是细胞质遗传D.线粒体遗传同样遵循基因的分离规律E.线粒体遗传的表现度与突变型mtDNA的数量有关。
3.以下符合mtDNA结构特点的是______.A.全长61569bpB.与组蛋白结合 C。
呈闭环双链状D.重链(H链)富含胞嘌呤E.轻链(L链)富含鸟嘧啶4.人类mtDNA的结构特点是______。
A. 全长16.6kb,不与组蛋白结合,为裸露闭环单链B. 全长61。
6kb,不与组蛋白结合,分为重链和轻链C。
全长16.6kb,与组蛋白结合,为闭环双链D. 全长61。
6kb,不与组蛋白结合,为裸露闭环单链E. 全长16。
6kb,不与组蛋白结合,为裸露闭环双链5.下面关于mtDNA的描述中,不正确的是______。
A.mtDNA的表达与核DNA无关B.mtDNA是双链环状DNAC.mtDNA转录方式类似于原核细胞D.mtDNA有重链和轻链之分E.mtDNA的两条链都有编码功能6.线粒体遗传属于______。
A.多基因遗传 B。
显性遗传C.隐性遗传D.非孟德尔遗传 E。
体细胞遗传7. 线粒体中的tRNA兼用性较强,tRNA数量为______。
A.48个B.32个C.64个 D。
61个 E.22个8.mtDNA编码线粒体中______。
A。
全部呼吸链-氧化磷酸化系统的蛋白质 B. 约10%的蛋白质C. 大部分蛋白质 D。
线粒体基质中的全部蛋白质E。
线粒体膜上的全部蛋白质9。
目前已发现与mtDNA有关的人类疾病种类约为______.A. 100余种B. 10多种C. 60多种 D。
线粒体遗传病ppt课件
1
一. 线粒体是动物细胞核外惟一含DNA的 细胞器。
二. 人类细胞线粒体DNA( mitochondrial DNA,mtDNA) 是人类基因组的组成部分,被称为“ 第25号染色体”。
三. 线粒体DNA突变引起线粒体遗传病。
四. 现已2 确认100多种mtDNA致病点突
第一节 线粒体基因组
16
(六)阈值效应
突变mtDNA达到一定数量才引起某种组织或器官 的功能异常。能影响能量代谢、引起特定组织或器 官功能障碍的最小量的mtDNA突变称为阈值。 mtDNA突变所致的特定组织或器官功能障碍表型 的出现,与某种组织野生型与突变型mtDNA的相 对比例有关。
17
影响阈值的因素:
1.组织对能量的依赖程度 不同组织和器官对能量的依赖程度不同,脑、骨骼肌、 心脏、肾脏、肝脏依次降低,说明脑组织阈值最低 。
15
(五)同质性与异质性
异质性(heteroplasmy):同一细胞或同一组织 中mtDNA分子上某一基因既有野生型,又有突变 型,称为异质性。此细胞或组织称为异质(野生型/ 突变型)。 同质性(homoplasmy):一个细胞或一种组织的 所有mtDNA分子上的某一基因都是相同的,称为 同质性。或均为野生型,或均为突变型。此细胞或 组织称为同质。 个体是否体现突变基因的表型,取决于突变基因的比 例、突变基因所在细胞的类型。
2. mtDNA突变的类型 不同的mtDNA基因突变,其阈值大小不同,tRNA基 因点突变:阈值为90%;大片段缺失阈值为60% 。
3.个体发育的阶段 同一器官,不同发育阶段,对能量依赖程度不同,因而 ,阈值不同。
4.细胞核的遗传背景
18
(七)母系遗传(maternal inheritance)
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
mtDNA与nDNA不同:
(1)其分子上无核苷酸结合蛋白,缺少组蛋 白的保护。 (2)线粒体内无DNA损伤修复系统,mtDNA易 发生突变并容易得到保存。 (3)每个线粒体内含有2~10个拷贝的mtDNA 分子。 (4)每个细胞可具有数千个mtDNA分子 。
二、线粒体DNA的遗传学特征
1. mtDNA半自主性 2. mtDNA遗传密码与通用密码不完全相同 3. mtDNA为母系遗传 4. mtDNA在有丝分裂和减数分裂期间都要经过 复制分离 5. mtDNA具有阈值效应的特征 6. mtDNA的突变率很高
心肌病
(一)Leber遗传性视神经病
(OMIM # 535000)
Leber T 医生
Leber遗传性视神经病( LHON)是由德国眼科 医生Theodor Leber在1871年首次报道的。本 病为母系遗传,存在性别差异,男女比例约 为4∶1。发病年龄通常在20~30岁,平均27 岁,有的病例最早在6岁发病,最迟在70多岁。 主要症状为急性或亚急性的双眼视力减退视 物模糊,多为双眼同时发病,有些病例双眼 先后相差1~6个月发病。由于视神经的坏死, 使得双眼的中央视力迅速丧失,但周围视力 仍存在。患者可能还有周围神经退化、震颤、 心脏传导阻滞和行动异常等表现。
I
1 2
II
1
2
3
4
5
6
III
1
2
3
4
5
6
7
Leber遗传性视神经病家系之二
Leber遗传性视神经病家系之三
Leber遗传性视神经病家系之四
(二)神经病伴运动性共济失调和视网膜 色素变性(OMIM # 551500)
以发育迟缓,近端肢体肌无力,痴呆, 抽搐,视网膜色素变性和感觉功能减退为特 点。 在线粒体ATPase6基因的第8993位点发 生T至G的颠换,使ATPase 第6亚单位的第 156位上的亮氨酸改变为精氨酸。
在MELAS患者中,异常的线粒体不能够代谢 丙酮酸,导致大量丙酮酸生成乳酸,而后者在 血液和其他体液中累积。MELAS患者的特征性 病理变化就是在脑和肌肉的小动脉和毛细血管 管壁中有大量的形态异常的聚集的线粒体。 超过80%的突变率发生在tRNAleu(UUR)基因上 的A3243G突变。
MTTL1*MELAS3243G
人的细胞里通常有上千个mtDNA拷贝,
在突变体和正常mtDNA共存的细胞中, mtDNA在细胞的复制和分离过程中发 生遗传漂变,可导致子细胞出现三种 基因型:纯合的突变体mtDNA、纯合 的正常mtDNA、突变体和正常的mtDNA 的杂合体。
(五)mtDNA具有阈值效应的特性
同质性是指同一组织或细胞中具有相同mtDNA的
线粒体病的常见临床表现 神经系统表现
脑血管意外样症状
癫痫发作
感觉性神经耳聋
共济失调
眼肌麻痹
肌阵挛
健忘
外周性神经病
眼神经病
肌病 抑制性精神病
血管性头痛
脊髓病
其他系统疾病表现
心脏传导系统缺陷 范可尼近端神经元机能障碍 视网膜色素变性 肾小球病肝病 小肠假性梗阻 糖尿病 个体矮小 乳酸中毒 全血减少
如果8993位臵突变的 异质性超过90%时,通常会 发生一种致命的、发生在 婴儿期的疾病,称Leigh综 合征(Leigh syndrome, LS,OMIM # 256000)。主 要病理学特征是基底神经 节和脑干部位神经元细胞 的退化 。
Leigh综合征患者
NARP和Leigh综合 征主要与ATP复合酶的功 能受损有关,目前发现 该病的致病突变主要是 mtDNA第8993位点 (ATPase6基因)T→G或 T→C,将Pase 6亚基156 位的亮氨酸改变为精氨 酸或脯氨酸,从而影响 ATP合酶的质子通道。
mtDNA结构 16569 bp
MELAS患者脑部病变
(五)链霉素耳毒性耳聋(OMIM # 580000)
1993年,Prezant等通过3个母系遗传的 氨基糖甙类抗生素致聋(aminoglycoside antibiotics induced deafness,AAID)家系 的研究,首次报道mtDNA编码的12SrRNA基因 1555位点A→G的突变。
mtDNA结构
16569 bp
MTTK*MERRF 8344G
(四)MELAS综合征(OMIM # 540000)
40岁以前开始出现的复发性休克、肌病、 共济失调、肌阵挛、痴呆和耳聋。少数患者出 现反复呕吐、周期性的偏头痛、糖尿病。进行 性眼外肌无力或麻痹使眼的水平运动受限,眼 外肌麻痹,眼睑下垂。肌无力,身材矮小等。
终止密码
甲硫氨酸 甲硫氨酸
(三)mtDNA为母系遗传
母亲将她的mtDNA
传递给儿子和女儿,
但只有女儿能将其
mtDNA传递给下一代。
线粒体的母系遗传,O:卵子; S:精子; B、C子细胞; Z:受精卵
A、
(四)mtDNA在有丝分裂和减数分裂期间都 要经过复制分离
一个人的卵母中大约含有105个线粒体,但当卵 母细胞成熟时,绝大多数线粒体数目可能会少 于10个,最多不会超过100个。线粒体数目从 105个锐减至少于100个的过程称为遗传瓶颈。 如果通过瓶颈存活下来的一个线粒体恰巧携带 一种突变基因,那么这个基因组就能够确保该 线粒体类型在发育完成之后的个体中的数量。
现象,异质性是指同一细胞或同一组织中具有
不同mtDNA的现象。线粒体病发病有一阈值,
只有当异常的mtDNA超过阈值时才发病。女性
携带者的细胞内突变的mtDNA未达到阈值或在
某种程度上受核影响而未发病,但仍可以通过
mtDNA突变体向下代传递。
(六)mtDNA的突变率极高
mtDNA中基因排列紧凑,任何突变都可能会影响
Complex Subunits Nuclear Ⅰ 41 34 Ⅱ 4 4 Ⅲ 11 10 Ⅳ 13 10 ATPase 14 12 合计 87 70 mtDNA 7 0 1 3 2 13
(二)线粒体基因组所用的遗传密码 和通用密码不完全等同
Codon UGG UGA AGG AGA AUG AUA nDNA 精氨酸 终止密码 精氨酸 精氨酸 甲硫氨酸 异亮氨酸 mtDNA 终止密码 终止密码 终止密码
(一)mtDNA具有半自主性
线粒体是一种半自主细胞器,受线粒体基 因组和核基因组两套遗传系统共同控制 。 线粒体DNA能够编码自己的mRNA、rRNA和 tRNA,合成一部分自身所需的蛋白质,线粒 体的这一功能称为线粒体的半自主性。线粒 体中的大多数蛋白质是核基因编码的,在细 胞质中合成。因此,线粒体的生长繁殖是核 -质两套遗传系统共同控制的结果
在这类突变中,当突变的mtDNA不超过85%时, 不发病,一旦超过这一水平,就会表现出严重 的临床症状。同时随年龄增长从不表现症状, 逐渐加重,直至完全表现,比错义突变的疾病 表现出更具系统性特征。许多神经肌肉性疾病 及心脏、肾脏等病变与此类突变相关,典型疾 病包括有线粒体脑肌病、线粒体肌病、肌阵挛 性癫痫伴破损性红肌纤维病、感觉神经性耳聋 等。
第六章 线粒体遗传病
( disease of mitochondrial inheritance)
1894年, Altmann发现动物细胞中线粒体。 1963年,M.Nass和S.Nass首次在鸡卵母细胞中 发现线粒体中存在有DNA。
1987年,Wallace等通过对线粒体DNA突变和
Leber病的关系的研究后,明确地提出线粒体
mtDNA仅编码13种蛋白质,绝大部分蛋白 质亚基和其他维持线粒体结构和功能的蛋白质 都依赖于nDNA编码,在细胞质中合成后,经特 定转运方式进入线粒体 。
mtDNA基因的表达受nDNA的制约,线粒体 氧化磷酸酶化系统的组装和维护需要nDNA和 mtDNA的协调,二者共同作用参与机体代谢调 节。
线粒体的半自主性
另一个导致听力丧失的纯质性突变是 7445位点T→G的突变 。
DEAF1555位点A→G
mtDNA结构 16569 bp
7445位点T→G
氨基糖甙类抗生素耳神经毒性
(六)KSS(OMIM # 530000)( CPEO)
常见临床表现是进行性眼外肌麻痹 (PEO)和视网膜色素变性。还包括心肌电传 导异常,共济失调,耳聋,痴呆和糖尿病。 发病年龄一般低于20岁,大多数患者在确诊 后几年内死亡。
mtDNA: 双链环状的DNA分子、裸露不与 组蛋白结合,分散在线粒体基质中,长约5μm、 含16569个碱基对。 12种多肽链 12S rRNA 外环为重链(H)富含G 16S rRNA 14种tRNA 内环为轻链(L)富含C 1种多肽链 8种tRNA
mtDNA共有37个基因
2种 编码 rRNA(12S和16S) 基因 22种 编码 tRNA 基因 13种 编码 蛋白 质基因
KSS并不表现出特定的母系或核基因遗
传方式。然而症状表明是一种线粒体病。
KSS患者的线粒体分析表明有mtDNA结构
第一节
线粒体DNA的结构特 点与遗传特征
一、 线粒体DNA的结构特点
ComplexⅠ ComplexⅢ
mtDNA结构 16569 bp
ቤተ መጻሕፍቲ ባይዱ
ComplexⅣ ATPase 8/6 rRNA tRNA
mtDNA分子量小,结构简单,进化速度 快,无组织特异性,具有特殊的结构特 征、遗传特征和重要功能,而且细胞中 含量丰富(几乎每个人体细胞中都含有 数以百计的线粒体,一个线粒体内有 2~10个拷贝的DNA),易于纯化,是研 究基因结构和表达、调控的良好模型, 在人类学、发育生物学、分子生物学、 临床医学、法医学等领域受到广泛的重 视,并取得令人瞩目的成就。
2.缺失、插入突变
mtDNA缺失突变引起绝大多数眼肌病, 这种缺失导致的疾病一般无家族史。