基因表达谱公共数据库
查基因cdna序列

查基因cdna序列概述在分子生物学和基因组学研究中,查找基因的cDNA序列是非常重要的一步。
cDNA (complementary DNA)是由利用反转录酶将mRNA转录成的DNA。
通过查找基因的cDNA序列,我们可以获取与该基因相关的信息,包括基因的序列、结构、功能等。
为什么要查找基因cDNA序列查找基因cDNA序列的主要目的是了解基因的功能以及它在生物系统中的作用。
通过查找基因cDNA序列,我们可以找到基因编码的蛋白质序列,进一步推测蛋白质的结构和功能。
此外,还可以利用cDNA序列来做基因表达的分析,了解基因在不同组织和生理状态下的表达模式。
查找基因cDNA序列的方法目前,有多种方法可以用来查找基因的cDNA序列。
下面是常用的几种方法:公共数据库公共数据库如NCBI的GenBank是查找基因cDNA序列的重要资源。
这些数据库中存储了大量的基因序列和相关信息,包括已知的人类基因组、动植物基因组等。
通过在这些数据库中进行搜索,我们可以找到已经注释好的基因的cDNA序列。
基因注释工具基因注释工具如NCBI的BLAST、Ensembl和UCSC等可以通过比对已知的基因组序列来预测基因的cDNA序列。
这些工具可以根据核酸或氨基酸序列进行比对,通过比对结果来推测基因的结构和序列。
实验方法如果需要获取特定基因的cDNA序列,一种方法是通过实验来获取。
常用的实验方法包括逆转录聚合酶链式反应(RT-PCR)和快速扩增cDNA末端(RACE)等。
查找基因cDNA序列的应用查找基因cDNA序列在许多研究领域都有广泛的应用。
下面是几个常见的应用场景:基因功能研究通过查找基因的cDNA序列,可以了解基因的编码蛋白质的结构和功能。
这对于研究基因的生物学功能非常重要,可以帮助揭示基因在生物系统中的作用机制。
基因家族分析通过查找基因的cDNA序列,可以发现存在于不同物种中的相似基因。
这些相似基因组成了基因家族,对于研究基因家族的起源和进化具有重要意义。
geo数据挖掘生信技能树简书

geo数据挖掘生信技能树简书geo数据挖掘生信技能树简书近年来,随着生物信息学的迅速发展,越来越多的数据产生和积累。
生物信息学家们如何从海量的生物数据中挖掘出有意义的信息,成为了一个重要课题。
而地理调查和数据挖掘(GEO)是一种非常有效的方法,可以帮助我们发现生物体与地理环境之间的关联性。
在这篇文章中,我们将介绍如何利用GEO进行数据挖掘以及相关的生信技能树。
GEO数据是指在GEO数据库中存储的大量生物测序数据,包括基因表达谱、染色质结构和修饰以及其他与基因组有关的信息。
这些数据被广泛应用于研究细胞、组织、器官和生物体的功能和发展过程。
然而,在海量的GEO数据中找到有用的信息并不是一件容易的事情。
首先,要想进行GEO数据挖掘,我们需要掌握一些基本的生信技能。
这包括熟练运用Linux操作系统和命令行工具,掌握编程语言如Python和R,并了解常用的生物信息学工具和数据库。
此外,基本的统计学知识也是必不可少的,因为在数据挖掘过程中我们需要对数据进行统计分析。
其次,了解GEO数据库和其相关的软件工具也是非常重要的。
GEO 数据库是由美国国家生物技术信息中心(NCBI)维护的一个公共数据库,其中包含了来自全球各地的生物信息数据。
掌握GEO数据库的使用方法,可以帮助我们快速地找到所需的数据,并进行后续的分析。
在GEO数据挖掘过程中,我们通常需要进行下述步骤:数据获取、数据预处理、数据分析和结果解释。
首先,我们需要根据具体的研究目的,在GEO数据库中检索并下载所需的数据集。
然后,对数据集进行质量控制和预处理,包括去除噪声和异常值,并将数据转换为适合后续分析的格式。
接下来,我们可以利用统计学和生物信息学的方法对数据进行分析。
常用的方法包括差异表达分析、聚类分析和功能富集分析等。
通过这些方法,我们可以找到与研究主题相关的差异表达基因、蛋白质或其他生物分子,并探索它们之间的功能和相互关系。
最后,我们需要解释和可视化分析结果。
生物信息学表达谱流程简介

DGEII
GO功能显著性分析结果文件:
DGEII
2.12、Pathway显著性富集分析 主要原理:在生物体内,不同基因相互协调行使其生物学,基 于pathway的分析有助于更进一步了解基因的生物学功能。KEGG是 有关pathway的主要公共数据库(Kanehisa, Araki, et al. 2008 )。Pathway显著性富集分析以KEGG Pathway为单位(对于非模式 物种,可以同blast比对来获得背景序列的KO号),应用超几何检 验,找出与整个基因组背景相比,在差异表达基因中显著性富集的 pathway(一般地,Qvalue≤0.05)。通过pathway显著性富集能确 定差异表达基因参与的代谢途径。
DGEII Pathway显著性富集分析结果:
各列的意义:
DGEII
DGEII 2.13、蛋白质相互作用网络分析 相互作用网络分析整合了 BIND,BioGrid,HPRD等相互 作用网络数据库的信息,结果 文件中的网络由差异表达基因 以及跟差异表达基因有直接相 互作用的基因组成。结果文件 可用Medusa软件显示。
DGEII
2.9、差异表达基因筛选
通过比较不同样本间的数据从而筛选出差异表达基因,后续分析中的差异基 因表达模式聚类分析,Gene Ontology功能显著性富集分析,Pathway显著性富集 分析,蛋白互作网络分析均是基于差异表达基因。 参照Audic S等人发表在Genome Research上的数字化基因表达谱差异基因检 测方法(Audic and Claverie 1997)(该文献已被引用超过五百次),我们开发了严 格的算法筛选两样本间的差异表达基因。 假设观测到基因A对应的一小部分,在这种情况下,p(x)的分布服从泊松分 布:
研究miRNA,这些数据库你必须得知道!常用数据库汇总

研究miRNA,这些数据库你必须得知道!常⽤数据库汇总miRNA是近年来在多种真核细胞及病毒中发现的⼀类来源内源性染⾊体上的⾮编码单链RNA,长度为21~25nt的短序列,在进化上具有⾼度的保守性,能够通过与靶mRNA特异性的碱基互补配对,引起靶mRNA降解或者抑制其翻译,从⽽对基因进⾏转录后的表达调控。
miRNA由⼀段具有发夹环结构的长度为70~80个核苷酸的miRNA前体(pre-miRNA)剪切后⽣成。
它通过与其⽬标mRNA分⼦的3′端⾮编码区域(3-untranslated region, 3′ UTR)互补导致该mRNA分⼦的翻译受到抑制。
最先发现的miRNAs是线⾍中控制发育时序的lin-4和let-7基因。
现已发现miRNA⼴泛地存在哺乳动物、线⾍、果蝇和植物等⽣物中。
除了lin-4和let-7基因外,其他miRNAs现在统⼀⽤miR-#表⽰miRNA,同⼀物种内相同或极相近似的miRNA可以使⽤相同的数字,只是进⼀步在数字之后加数字或字母作为后缀以区别其基因在序列上只有微⼩的差别。
尽管miRNA基因不编码蛋⽩质,但其编码的RNA在⽣物的整个⽣命过程中发挥着重要作⽤。
miRBasemiRBase数据库是⼀个提供包括已发表的miRNA序列数据、注释、预测基因靶标等信息的全⽅位数据库,是存储miRNA信息最主要的公共数据库之⼀。
该数据库于2014年6⽉更新为最新版本V21.0,包含223个物种的35828个成熟的miRNA序列。
该数据库提供便捷的⽹上查询服务,允许⽤户使⽤关键词或序列在线搜索已知的miRNA和靶标信息(仅包含已有的靶标信息,所以会出现部分miRNA靶标信息⽆的现象)。
该数据库⽤于miRNA信息查询较多,靶关系预测较少。
TargetScanTargetScan数据库是⼤家⽐较常⽤的预测miRNA靶基因数据库,主要通过搜索和每条miRNA种⼦区域匹配的保守的8mer和7mer位点来预测靶基因。
基于GEO_和TCGA_数据库对肺腺癌差异表达基因的生物信息学分析

第 49 卷第 6 期2023年 11 月吉林大学学报(医学版)Journal of Jilin University(Medicine Edition)Vol.49 No.6Nov.2023DOI:10.13481/j.1671‑587X.20230612基于GEO和TCGA数据库对肺腺癌差异表达基因的生物信息学分析叶汇, 孙哲, 周丽婷, 齐雯, 叶琳(吉林大学公共卫生学院劳动卫生与环境卫生教研室,吉林长春130021)[摘要]目的目的:采用生物信息学方法筛选影响肺腺癌(LUAD)的关键基因,分析其生物学功能及其对LUAD预后的影响。
方法方法:于高通量基因表达(GEO)数据库下载GSE118370和GSE136043芯片数据,癌症基因组图谱(TCGA)数据库筛选LUAD相关数据。
采用R软件分析共同表达的差异表达基因(DEGs)。
采用clusterProfile R包对DEGs进行基因本体(GO)功能富集分析,DAVID数据库进行京都基因与基因组百科全书(KEGG)通路富集分析,STRING数据库构建蛋白-蛋白相互作用(PPI)网络。
采用Cytoscape筛选连接度排名前10位的关键基因,GEPIA数据库和人类蛋白质图谱(HPA)数据库分析正常肺组织和LUAD组织中关键基因mRNA和蛋白表达情况及不同分期LUAD组织中关键基因表达情况。
关键基因免疫浸润分析和生存分析获取关键基因表达与患者生存期的相关关系。
结果:共筛选DEGs 428个。
GO分析,LUAD的DEGs在主要富集于上皮-间质转化(EMT)等生物过程(BP)方面、细胞基部等细胞组分(CC)方面和细胞外基质(ECM)结构形成等分子功能(MF)方面。
KEGG分析,LUAD的DEGs主要富集于细胞因子受体相互作用通路等方面。
筛选DNA拓扑异构酶Ⅱα(TOP2A)、果蝇纺锤体异常基因(ASPM)、细胞周期蛋白B1(CCNB1)、人类细胞分裂周期相关基因8(CDCA8)、含杆状病毒IAP重复序列蛋白5(BIRC5)、苏氨酸激酶(AURKA)、驱动蛋白超家族成员20A(KIF20A)、中心体相关蛋白55(CEP55)、着丝粒蛋白F(CENPF)和微管组织因子(TPX2)为关键基因。
单细胞数据提取注释信息

单细胞数据提取注释信息是单细胞RNA测序(single-cell RNA-sequencing,scRNA-seq)数据分析的一个重要步骤。
在单细胞RNA测序实验中,通常会获得大量的单细胞数据,每个数据点代表一个细胞的基因表达水平。
为了更好地理解这些数据,需要对每个细胞进行注释,以提供关于细胞类型、细胞状态、细胞来源等信息。
以下是一些常用的方法来提取和注释单细胞数据:
1. 细胞类型注释:
通过已知的细胞标记基因来识别和注释细胞类型。
例如,可以通过与已知细胞类型相关的基因表达模式来识别神经细胞、肌肉细胞等。
2. 细胞状态注释:
对于处于不同分化阶段或不同生理状态的细胞,可以通过检测特定基因的表达水平来判断其分化阶段或生理状态。
3. 细胞来源注释:
在多细胞生物中,不同组织或器官的细胞可能具有独特的基因表达模式。
通过比较细胞表达谱与已知组织或器官的特异性基因表达模式,可以推断细胞的来源。
4. 分子途径分析:
通过分析细胞中特定信号通路或代谢途径的相关基因表达水平,可以推断细胞的功能和特性。
5. 聚类分析:
通过对细胞的基因表达谱进行聚类分析,可以将具有相似基因表达模式的细胞分为一组,从而揭示细胞的相似性和差异性。
6. 整合外部知识:
利用公共数据库中的细胞类型特异性基因标记和知识,可以对单细胞数据进行注释。
例如,可以利用CellMarker、CellNet等数据库来识别和注释细胞类型。
7. 使用生物信息学工具:
利用各种生物信息学工具和软件,如Seurat、Scanpy等,可以帮助分析单细胞数据,并提取有用的注释信息。
常用的生物信息学软件的介绍和文献依据
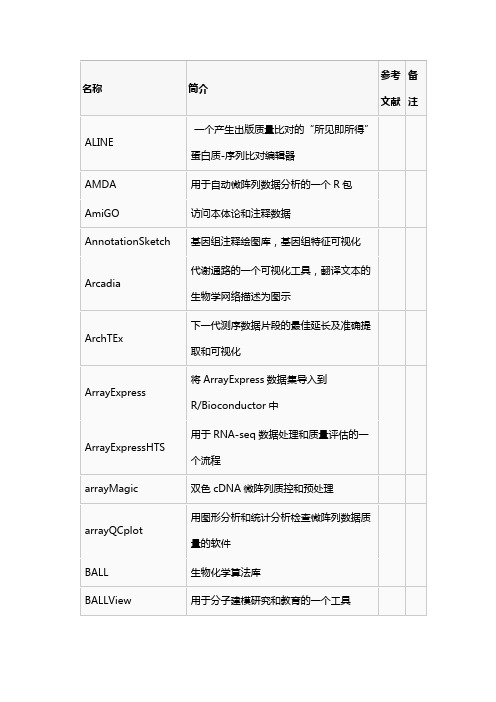
BioWarehouse
一个生物信息学数据仓库整合工具包
birgHPC
为生物信息学和分子动力学创建即时计算集群,自启动linux发行版
Biskit
python编写的一个结构生物信息学软件平台(库)
BisoGenet
一个新的基因网络构建、可视化和分析工具,cytoscape插件
一个促进高通量测序分析的基于云计算的框架
ESBTL
用于生物大分子结构和几何分析的高效PDB剖析器和数据结构
Expander
一个整合的基因表达数据分析软件平台,支持微阵列数据
分析的所有阶段
ExpressionPlot
一个分析RNA-Seq和微阵列基因表达数据的基于网络的框架
EZ-Viz
用标签和按钮简化PyMOL中分子查看
ChIPpeakAnno
一个注释ChIP-seq和ChIP-chip数据(峰)的Bioconductor包
ChIPseqR
核小体定位和组蛋白修饰ChIP-seq实验分析
Chipster
用于微阵列和其他高通量数据的用户友好的分析软件
CisGenome
一个分析ChIP-chip和ChIP-Seq的整合软件系统
病毒的传播和重组事件
J-Express
使用Java来探索基因表达数据
Jalview
Java多重序列比对编辑器
Java Treeview
微阵列数据可视化,树状图查看
JBrowse
下一代基因组浏览器,通过平滑地动态移动,缩放,导航基因组注释
jClust
一个聚类和可视化工具箱
JColorGrid
生物学测量值可视化,绘制热图,颜色网格等
植物miRNA基因组学数据库构建及intronic

五、应用实例
五、应用实例
为了展示植物miRNA基因组学数据库的功能和优势,以下给出一个应用实例。 某研究团队需要研究一种重要植物miRNA在响应环境刺激中的作用。通过使用 本次演示构建的植物miRNA基因组学数据库,该团队成功地查询到了这种植物 miRNA在不同环境条件下的表达谱数据,
四、比较基因组学研究
四、比较基因组学研究
通过对不同植物物种的细胞器基因组进行比较基因组学研究,我们可以更好 地理解植物的进化历程和生物学特性。例如,通过比较不同物种的叶绿体基因组, 我们可以看到这些基因组的共性和差异,从而理解叶绿体在进化过程中的变化和 保留。同样,通过比较不同物种的线粒体基因组,我们可以理解线粒体在植物进 化过程中的作用和变化。
谢谢观看
3、查询引擎:提供高效的查询引擎,支持多种查询方式,如序列查询、基因 组位置查询、功能分类查询等。
三、功能模块
三、功能模块
植物miRNA基因组学数据库应具备以下功能模块: 1、用户界面:提供简洁明了的用户界面,方便用户输入查询条件、展示查询 结果以及对数据库进行管理。
三、功能模块
2、系统管理:支持管理员对数据库进行管理,包括用户管理、数据更新、系 统维护等。
一、背景介绍
一、背景介绍
植物miRNA基因组学研究的意义在于:一方面,揭示植物生长和发育的奥秘, 为农业生产提思 路,为植物基因工程提供新的工具和资源。为了满足科研人员对植物miRNA数据 的需求,构建一个专门的植物miRNA基因组学数据库是必要的。
参考内容
标题:植物细胞器基因组测序、 组装及比较基因组学研究
一、引言
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
基因表达谱公共数据库 (2010-04-17 11:17:21)转载▼ 标签: 教育 分类: 系统生物学 gene expression profiles: NCBI: GEO EBI ArrayExpress 和 SMD
功能基因组相关信息分析 功能基因组学是后基因组研究的核心内容,它强调发展和应用整体的(基因组水平或系统水平)实验方法分析基因组序列信息阐明基因功能,特点是采用高通量的实验方法结合的大规模数据统计计算方法进行研究,基本策略是从研究单一基因或蛋白上升到从系统角度一次研究所有基因或蛋白。随着功能基因组实验研究的深入,大量的数据不断涌现,生物信息学将在功能基因组学研究中的扮演关键角色。 7.1 大规模基因表达谱分析 随着人类基因组测序逐渐接近完成,科学家发现即使获得了完整基因图谱,对了解生命活动还有很大距离。我们从基因图谱不知道基因表达的产物是否出现与何时出现;基因表达产物的浓度是多少;是否存在翻译后的修饰过程,若存在是如何修饰的,等一系列问题。这些问题的实质是不了解按照特定的时间、空间进行的基因表达谱。获得基因表达的信息是比DNA序列测定艰巨得多的任务,因为基因表达是依赖于许多因素的动态过程。 国际上在核酸和蛋白质两个层次上发展了分析基因表达谱的新技术,即核酸层次上的 cDNA 芯片(cDNA微阵列)技术和蛋白质层次上的二维凝胶电泳和测序质谱技术,即蛋白质组(proteome)技术。DNA芯片技术能够在基因组水平分析基因表达,检测许多基因的转录水平。 对大规模基因表达谱的分析存在新的方法学问题,它们从数学角度看不是简单的NP问题、动力系统问题或不确定性问题,而是基因表达网络,因此需要发展新的方法和工具。同时,在芯片等的设计上,也需要从理论到软件的支持 下面主要围绕cDNA芯片相关的数据管理和分析问题进行讨论。 1.实验室信息管理系统 cDNA芯片实验的目的是要在一次实验中同时得到成千上万个基因的表达行为,这样的实验需要有管理实验前后大量数据的能力。设计构建检测基因表达的微阵列需要获得生物体基因的所有序列、注释和克隆。在杂交反应和扫描后,收集到的数据必须以某种方式保存,以便很容易进行图象处理和统计及生物学分析。因此需要建立与大规模高通量实验方法相匹配的实验材料和信息管理系统。 该系统除用来定位和跟踪材料来源(例如,克隆,微阵列,探针)外,还必须管理实验前后大量的数据。此外,还包括实验室设备软件系统,如斯坦福大学Brown实验室免费的控制自制机器点样设备软件(http://cmgm.standford.edu/pbrown) 芯片图象处理已有各种软件工具,基本的功能是将不同信号强度点的图像转换为每个点的强度数值。这方面没有一致的方法,许多研究小组仍在开发这类软件。图象分析软件的质量对精确解释玻片和膜上的信号非常关键。NHGRI的Yidong Chen开发了一种复杂的图象分析程序,deArray,可免费获取。 美国国立卫生研究院人类基因组研究所(NHGRI)开发的免费的cDNA芯片数据管理分析系统ArrayDB,涉及微阵列的设计、实验室信息管理、实验结果的处理和解释。下面加以简单介绍。 ArrayDB ArrayDB是用来储存、查询和分析cDNA芯片实验信息的实验室管理系统。ArrayDB整合了 cDNA芯片实验中的多个方面,包括数据管理、用户介面、机器自动点样、扫描和图象处理。ArrayDB中保存的数据包括实验来源、实验参数和条件以及原始的和经处理的杂交结果。ArrayDB依托的关系数据库储存了芯片上每个克隆的相关信息,包括基因的简单描述、GenBank号、IMAGE克隆识别号、代谢途径号和实验室内部克隆号。ArrayDB还储存了与cDNA芯片制造和实验条件的信息。包括点样相关数据(点样机器的参数)、环境条件(温度、湿度、点样针冲洗条件)等数据。此外,还保存了杂交探针和实验条件,包括研究者的姓名,研究目的和实验条件、组织细胞类型的文本描述。有关杂交的结果的信息包括扫描图象(“原始”结果)、信号强度数据、信号强度比值和本底值。 ArrayDB的设计允许灵活地提取数据信息。设计策略允许不同来源的数据输入,大多数克隆信息来自Unigene数据库(包括序列的命名和获取号)。也允许新分离的还没有获取号及名称的克隆的输入。许多数据输入和处理过程是自动的。软件会自动扫描目录查找新输入数据库中的信号强度数据无须人工辅助,其它自动处理包括很方便地整合信号强度数据和克隆数据。 ArrayDB的Web界面能很方便地进行不同类型信息的查询,从克隆信息到信号强度值到分析结果。ArrayDB支持各种字段的数据查询,例如克隆ID、标题、实验编号、序列获取号、微量滴定板编号以及相关克隆的结果。每个克隆的更多信息通过超文本链接至其他数据库如dbEST、GenBank或Unigene,代谢途径信息也可通过链接至KEGG得到。 通过序列相似性搜索可以有效地寻找目的基因。ArrayDB支持对10K/15K数据(软件自带数据)进行BLASTN搜索以便确定目的基因是否已包含在芯片中。 ArrayDB能分析单个和多个实验产生的信号强度比值的类型和关系。ArrayViewer工具支持查询和分析单个实验;MultiExperiment viewer工具支持多个实验数据。在下述网站可得到更详细信息和相关软件。 DeArray和ArrayDB网址: http://www.nhgri.nih.gov/DIR/LCG/15K/HTML
基因表达公共数据库 数据库用途 (1)基础研究将来自各种生物的表达数据与其它各种分子生物学数据资源,如经注释的基因组序列、启动子、代谢途径数据库等结合,有助于理解基因调控网络、代谢途径、细胞分化和组织发育。例如,比较未知基因与已知基因表达谱的相似性能帮助推测未知基因的功能。 (2)医学及药学研究例如,如果特定的一些基因的高表达与某种肿瘤密切相关,可以研究这些或其它有相似表达谱的基因的表达的影响条件,或研究能降低表达水平的化合物(潜在药物)。 (3)诊断研究 通过对数据库数据进行基因表达谱的相似性比较对疾病早期诊断具有临床价值。 (4)毒理学研究 例如,了解大鼠某种基因对特定毒剂的反应可帮助预测人的同源性基因的反应情况。 (5)实验质量控制和研究参考实验室样本与数据库中标准对照样本比较能找出方法和设备问题。此外,还能提供其他研究者的研究现状,避免重复实验,节约经费。 数据库的特点和难点 目前急需建立标准注释的公共数据库,但这是生物信息学迄今面临的最复杂且富有挑战性的工作之一。主要困难来自对实验条件细节的描述,不精确的表达水平相对定量方法以及不断增长的庞大数据量。 目前所有的基因表达水平定量都是相对的:哪些基因差异表达仅仅是与另外一个实验比较而 言,或者与相同实验的另一个基因的相比而言。这种方法不能确定mRNA的拷贝数,转录水平是总的细胞群的平均水平。结果导致采用不同技术进行基因表达的检测,甚至不同实验室采用相同技术,都有可能不能进行比较。对不同来源数据的进行比较有必要采取两个步骤:首先,原始数据应避免任何改动,比如采取数据标准化(data-normalization)的方法。其次,在实验中设计使用标准化的对照探针和样本以便给出参考点至少使来自同一实验平台的数据标准化。 另一难点是对实验条件的描述,解决方法是对实验方法用采用规范化词汇的文件描述:如基因名称,物种,发育阶段,组织或细胞系。还要考虑偶然的不受控制实验因素也可能影响表达:例如空气湿度,甚至实验室的噪音水平。目前建立一种结构能对将来实验设计的所有细节进行描述显然是不可能的。比较现实的解决办法是大部分采用自由文本描述实验,同时尽可能加上有实用价值的结构。DNA芯片实验的标准注释必须采用一致的术语,这有待时间去发展。但目前,就应采用尽可能合理的标准用于DNA芯片数据及其注释。 标准化的基因表达公共数据库要有五类必要的信息: (1)联系信息:提交数据的实验室或研究人员的信息。 (2)杂交靶探针信息:对阵列上的每个“点”,应有相应的DNA序列在公共数据库中的编号。对cDNA阵列,克隆识别号(如IMAGE clone_id)应给出。 (3)杂交样本:细胞类型和组织来源用标准语言描述。常规诊断病理中使用的组织和组织病理词汇可被采用,还可采用胚胎发育和器官发生中的标准词汇。样本来源种属的分类学名称(如Saccharomyces cerevisiae,Homo sapiens),应当提供。对有些生物体如啮齿类动物和微生物,品系资料需要提供。关于实验中生物体状况的资料,如用药或未用药非常关键,也需提供。“肿瘤与正常”或不同发育阶段也该注明。细胞或生物体的遗传背景或基因型在特定例子中也应是重要的,如酵母基因缺失和转基因鼠。最后,由于组织处理的会引起差别,故应包括相关的详细处理方法。 (4)mRNA转录定量:这方面非常关键,很难通过一组“持家基因”做内参照进行标准化,有关的具体定量方法应提供。 (5)统计学意义:理想地,应经济合理地有足够的次数重复一个实验以便给出基因表达测定的变异情况,最好能提供合理的可信度值。 上述表达数据记录的前两个要求是简单的,第三个要求较困难需有标准术语协议,但这并不只是表达数据的要求,类似的要求已在公共序列数据库或专业化的数据库中得到成功解决。目前基因表达数据最富有挑战性的方面是最后两个方面。 现状和计划 几个大的芯片实验室如斯坦福大学和麻省理工学院Whitehead研究所等,在发展实验室内部数据库;大的商业化芯片公司如Affymetrix, Incyte,GeneLogic,正在开发基于Affymetrix芯片技术平台的商业化基因表达数据库。哈佛大学已经建立了一个的数据库,数据来自几个公共来源并统一格式。宾夕法尼亚大学计算生物学和信息学实验室正在整合描述样本的术语。 目前至少有3个大的公共基因表达数据库项目:美国基因组资源国家中心的GeneX;美国国家生物技术信息中心(NCBI)的Gene Expression Omnibus;欧洲生物信息学研究所(EBI)的ArrayExpress. 欧美专家合作提出有关数据库的初步标准:实验描述和数据表示的标准;芯片数据XML 交换格式;样本描述的术语;标准化、质量控制和跨平台比较;数据查询语言和数据挖掘途径。(http://www.ebi.ac.uk/microarray/)。EBI与德国癌症研究中心正在开发ArrayExpress , 一种与目前推荐标准兼容的基因表达数据库。该数据库将利用来自合作方的的数据,可操作的数据库将于近期建立(http://www.ebi.ac.uk/arrayexpress)。