肾钙质沉淀症

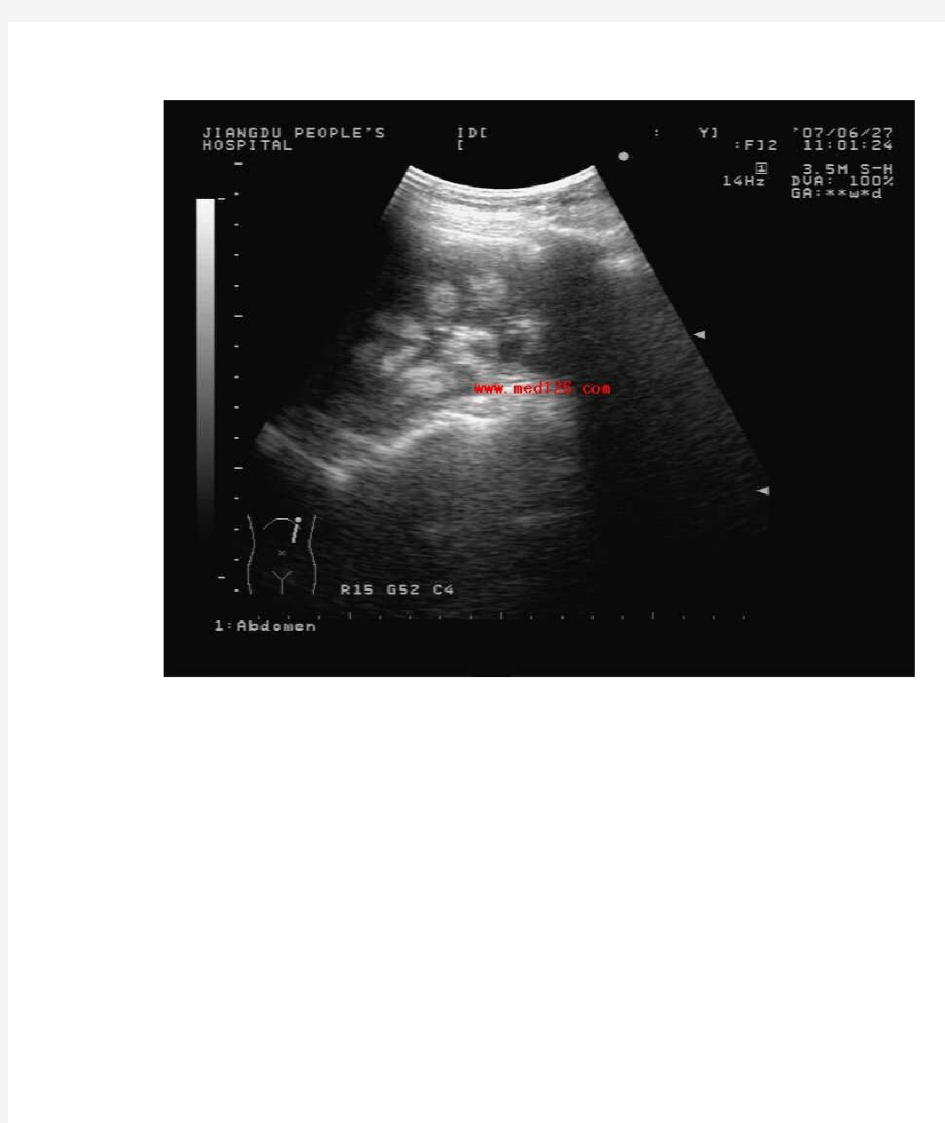
肾钙质沉淀症:
是钙质在肾组织内沉着。常为系统性、全身性疾病所致,所以为双侧肾发病,多发生于高血钙症,在甲状旁腺机能亢进、高氯血症性酸中毒和慢性肾盂肾炎时可见到,也见于醛固酮增多症、肾小管酸中毒等。肾钙质沉淀症X-线平片一般不显影,严重者可显影。但在超声检查时声像图极为清晰典型,双肾各锥体均完整显示为强回声,但无声影。肾钙质沉淀症在发病初始,仅在肾皮质与髓质交界处的肾髓质边缘出现一圈高回声,随着病情进展,高回声渐向肾髓质方向增宽,最后肾锥体的大部分直至全部为高回声所占据。
肾钙质沉淀症均双侧同时出现,且各锥体的病情演变进程基一致
肾萎缩概述
肾萎缩概述 肾萎缩atrophy of kidney是根据肾脏体积与人的年龄、性别和人体的身高、体重相互比较的,相对偏小或明显缩小的一种异常现象。长时间的肾脏疾病,导致肾单位,尤其肾小球受损,使整个肾脏出现体积缩小。肾萎缩和肾功能改变是相辅相成的,当患者出现肾萎缩时已经或者必然会出现肾功能不全及至肾功能衰竭--尿毒症。因此在治疗肾病过程中改善和控制肾萎缩是至关重要的。 何谓肾萎缩? 肾萎缩是一个病理解剖学名词,也就是说肾脏萎缩,体积明显减小,也称终末期肾脏。这时肾萎缩患者的肾小球、肾小管(即肾单位)已大部分或全部破坏,肾脏已失去生理功能。 通常对慢性肾衰患者进行肾脏B型超声检查,就可以了解肾脏是否患有肾萎缩,而且这种检查方法对患者没有损害。肾脏B超的正常值为:左肾长径8.1~11厘米,横径5.5~7厘米,厚度4~5厘米;右肾长径 7.9~10.5厘米,横径5.5~7厘米,厚度4~5厘米。 倘若测值明显小,说明已经患肾萎缩,处于末期,预后不佳,治疗效果也差。倘若测值略小或正常,说明 是中期或早期,治疗效果好,预后良。因而作为从事肾脏病的临床医生,了解患者肾脏大小,对分析病情、判 断预后、估计疗效均有十分重要的意义。 肾萎缩的原因 肾萎缩的病因应考虑为先天性肾发育不全或末期肾病的肾萎缩及肾血管疾病。肾萎缩可见于先天性肾发育不全 及有胶原和钙质沉着或某些急性病变,如:急性肾小球性疾病、糖尿病性肾硬变、肾移植排异、慢性肾小球肾 炎、肾皮质坏死、Alport综合征、急性肾小管坏死、高血压性肾硬化。局灶性或弥漫性肾实质解剖的破坏见于 任何肿块性病变(如:囊肿、肿瘤、脓肿和血肿),局灶性肾盂肾炎,实质瘢痕(如肾梗死或萎缩性肾盂肾炎), 婴儿型多囊肾,慢性肾盂肾炎或损伤等病变。肾血管性疾病所致肾萎缩可由以下几种疾病所致:动脉内膜疾患、 外伤、恶性病变或心脏疾病所致的栓塞。 单侧肾萎缩的临床分析 1 判断肾萎缩的标准 正常肾大小(长轴)左侧为11~13.5 cm(平均为12.2 cm),右侧为10.8~13 cm(平均为11.9 cm),相当于相邻三个腰椎体包括其椎间隙之和,宽度为长度的二分之一。一般认为两肾长度相差1.5 cm就有诊断意义,右肾小于左肾不到10%,多无肯定意义。肾影轻度缩小(<10~9 cm),中度缩小(9~8 cm),重度缩小(<8 cm)。 2 单侧肾萎缩临床特点 (1)对侧肾脏代偿性增大不常见,但肾血流量多数增强。 (2)单侧肾萎缩本身不引起高血压,如先天性肾发育不良、肾自截、肾结核、慢性肾盂肾炎不伴高血压,但肾动脉狭窄因激活肾素-血管紧张素-醇固酮系统而使血浆肾素水平升高,引起高血压常见。 (3)肾动脉狭窄伴患肢足背动脉搏动减弱或消失并不常见。 (4)肾动脉狭窄经DSA扩张术后,萎缩肾脏血流明显增加,肾脏较前明显增大,可见部分萎缩肾脏具有可逆性。 (5)肾动脉严重狭窄,探及不到血流,但通过侧枝循环使肾脏保持良好血供,不致病肾严重萎缩。 (6)在同位素肾图上,病肾显影延迟,排泄减慢,以排泄功能异常为先出现。肾血流明显减少或消失。 3 单侧肾萎缩临床意义 临床上单侧肾萎缩并不罕见,患者常以高血压、腰酸等症状就诊,行B超或KUB、IVP等检查时可发现。除原发病症状外,无明显血尿、蛋白尿、肾功能正常,但若在劳累、感染,特别是尿路感染、尿路梗阻、损害肾功能药物应用等诱发下,肾功能可能迅速恶化,若干年后双肾萎缩,应引起临床医师重视。 4.肾萎缩的检查 检查肾脏大小虽未能直接找到肾脏病的病因,但可初步了解病变的性质,提起医生的注意,或作急性、慢
巴特综合征-罕见病诊疗指南
103 巴特综合征 一、疾病概述 1962年,巴特等[1]首次报道了2例出现低钾性代谢性碱中毒、高醛固酮血症,血压正常,肾组织学检查显示肾小球旁器肥大的患者,遂将其命名为巴特综合征。巴特综合征属于罕见病,国外统计的发病率为0.001%[2],科威特发病率是1.7/100000活产婴儿,哥斯达黎加是1.2/100000活产婴儿,瑞典1. 2/1 000000人[3]。国内报道较少。巴特综合征(Bartter syndrome,BS)是指一组临床以低钾血症和代谢性碱中毒为特征的遗传性肾小管疾病。其遗传模式为常染色体隐性遗传,最主要的致病机制是NaCI在髓襻升支粗段和远曲小管的重吸收障碍。随着近代生物分子学技术的发展,根据致病基因的不同,目前,包括Ca2+受体基因变异在内,巴特综合征可以分为5个亚型(见图1)。新生儿I型Ⅱ型各自由于编码Na+-K+-2CI-协同转运体的SLC12A1基因的变异及编码内流K+通道的KCNJ1基因的变异而造成功能缺失。BSⅡ型是由于CLCNKB基因编码的CIC-Kb Cl-通道功能的丧失。BSⅡa型是由于编码Barttin蛋白,一种CIC-K凋解亚单位的BSND 基因的变异而造成的。Ⅱb型是一种双基因疾病.由于编码Cl-通道的CLCNKA 和CLCNKB基因的变异[4]。而编码基膜外侧的Ca2+敏感的CaSR基因的功能变异,导致了伴有甲状腺功能减退的BSV型。 NKCC2的缺乏导致了BSI型,这个蛋白是最主要的Na+,K+和Cl-从尿中重吸收入细胞的通路。ROMK的变异导致了BSⅡ型,再循环的K+无法由细胞回到肾小管管腔。患者在新生儿阶段表现为瞬间的高血钾。小管中K+的缺乏抑制了NKCC2。Cl-离开细胞通过CLCKNA和CLCKNB通道。CLCKNB通道的缺失属于BSⅡ型。BSⅡ型的患者缺乏Barttin产物,这是维持CLCKNA和CLCKNB 通道的正常功能,同样也表达于远端小管和内耳的蜗管。这也解释了儿童中的感音神经性耳聋。BSV型是由于钙敏感受体的获得性功能变异,导致了低钙,低钾和高尿钙。
Gitelman综合征
Gitelman综合征 Gitelman综合征(GS)是一种常染色体隐性遗传的失盐性肾小管疾病,临床特征为低钾代谢性碱中毒伴低镁血症和低尿钙症。 临床表现 多数GS患者于青少年或成年发病,但一些临床症状也可在儿童期甚至新生儿期出现,约1/3的患者可有明确的家族史。GS常见的临床症状多为非特异性,常与电解质紊乱及RAAS激活等有关,包括以下表现: 全身症状 肢体乏力、疲劳、运动耐量下降、口渴、多饮、嗜盐; 心血管系统 血压正常或偏低、心悸、QT间期延长、室性心律失常; 消化系统 发作性腹痛、便秘、呕吐;
泌尿系统 多尿、夜尿、遗尿、蛋白尿、低钾性肾病; 神经-肌肉系统 头晕、眩晕、共济失调、假性脑瘤、肢体麻木、感觉异常、肌肉痉挛、抽搐、横纹肌溶解; 骨关节系统 关节痛、软骨钙质沉着症; 生长发育 发育停滞、生长迟缓、青春期延迟。 需要指出,多数GS患者尿蛋白定量正常或轻度升高,一般为中小分子蛋白,可能与长期低钾所致的肾小管损伤有关,大多数患者肾功能正常,因此无需肾穿刺活检。但患者如果出现大量蛋白尿、原因不明的肾功能受损等,需行肾穿刺活检明确是否合并肾小球病变或其他肾
脏疾病。 实验室检查 生化及影像学检查 由于GS患者症状缺乏特异性,临床诊断更多依赖于实验室检查,典型患者临床表现为"五低一高"和代谢性碱中毒,即低血钾、低血镁、低血氯、低尿钙、偏低血压和RAAS活性增高。特别是低血镁和低尿钙对诊断GS有重要价值。支持GS诊断的实验室检查结果主要包括: ◆低钾血症及肾性失钾:血清钾<3.5 mmol/L(严重者<2.0 mmol/L,排除使用降钾类药物),常持续存在或反复出现,伴肾性失钾(尿钾/尿肌酐>2.0 mmol/mmol或血钾低于3.5 mmol/L时24 h尿钾>25 mmol); ◆代谢性碱中毒; ◆低镁血症及肾脏排泄镁增多:血镁<0.7 mmol/L,镁排泄分数(FEMg)>4%{FEMg=[尿镁(mmol/L)×血肌酐(mmol/L)]/[血镁(mmol/L)×尿肌酐(mmol/L)]}; ◆低尿钙:成人随机尿中尿钙/尿肌酐<0.2 mmol/mmol;
肾结石容易引发的几种并发症
肾结石容易引发的几种并发症 *导读:很多人明明知道自己患上肾结石了,但是却会犹豫 着要不要进行治疗,因为患者还会存有一定的侥幸心理,认为自己不…… 很多人明明知道自己患上肾结石了,但是却会犹豫着要不要进行治疗,因为患者还会存有一定的侥幸心理,认为自己不会有什么事的。肾结石对患者的危害不大,只是因为患者的患病时间不长,但是患者要是延误病情的话,就会导致哪些方面更严重的病症呢? 1.泌尿系梗阻肾结石结石致泌尿系管腔内堵塞可造成梗阻 部位以上的积水。结石性梗阻常为不完全性梗阻,有的结石表面有小沟,尿液可沿小沟通过;有时结石虽较大,甚至呈铸状结石,但尿仍能沿结石周围流出,也可能在长时间内不引起积水,肾盂壁纤维组织增生变厚时,则扩张表现不明显。 肾结石发生梗阻由于发病缓急不同,其临床表现有很大差异。尽管最终均可引起肾盂积水,但临床不一定以肾盂积水为主要表现。肾盂积水有时无任何临床症状,部分病例直到肾盂积水达严重程度,腹部出现肿物和肾功能不全,甚至无尿时才被发现。 2.局部损伤小而活动度大的结石,对局部组织的损伤很轻,大而固定的鹿角状结石可使肾盏、肾盂上皮细胞脱落,出现溃疡、纤维组织增生、中性粒细胞和淋巴细胞浸润,以致纤维化。移行
上皮细胞长期受结石刺激后,可发生鳞状上皮细胞化生、甚至可引起鳞状上皮细胞癌,因此应做尿脱落细胞学检查。尽管尿脱落细胞异常不一定能使之确诊,但从中可获得尿路上皮细胞发生异常改变的提示。对于长期存在的肾盂或膀胱结石都要想到上皮细胞癌变的可能,手术时应取活体组织送快速冰冻切片检查。 3.感染有无感染对肾结石的治疗和防治有重要意义。尿路 感染病人临床表现为发热、腰痛、尿中出现脓细胞。尿培养有细菌时,应同时做药敏试验。 结石合并感染时,可加速结石的增长和肾实质的损害。在结石排出或取出前,这种感染很难治愈,可发生肾盂肾炎、肾积脓、肾周围炎、严重者甚至可发展为肾周围脓肿;与腹膜粘连后,可 穿破入肠管。显微镜下可见肾间质炎症,细胞浸润和纤维化,肾小管内有中性粒细胞和上皮细胞,后期出现肾小管萎缩和肾小球硬化。 4.肾功能不全肾结石在合并尿路梗阻时,尤其是双侧尿路 梗阻或在此基础上合并严重感染,病人可出现肾功能不全。当梗阻解除和(或)感染得到有效控制,部分病人。肾功能可好转或恢复正常。 判断肾功能的方法除检测血清尿素氮、肌酐和内生肌酐清除外,还可采用静脉肾盂造影术并根据造影剂排出的时间、浓度加以判断。B超虽可了解尿路扩张情况和肾实质的厚度,但判断肾功能较为困难。静态或动态核素扫描或摄像可提供有价值的线索。
肿瘤样钙质沉着症
肿瘤样钙质沉着症( tumoral calcino sis, TC) 是一种发生于大关节附近, 但并不累及关节滑膜的特发性软组织钙质沉着性疾病。TC是一种很少见的疾病, 又名瘤样钙化症、钙化性胶原溶解、臀石等。1899年由Duret首先描述,1943年由Inclan命名。本病发病原因不明, 可能与下列因素有关: 钙磷、胆固醇代谢异常; 关节附近胶原纤维对刺激所作出的反应性钙化; 遗传因素, 认为是一种常染色体显性遗传性畸形所致,与遗传相关的家族性肿瘤样钙盐沉着症(familial tumoral calcinosis,FTC)。FTC 包含2 种亚型,即分别由GALNT3 基因或SAMD9 基因突变所致,前者多伴有高磷酸血症,后者则无; 外伤因素, 为反复轻微损伤造成局部营养障碍而引起。其他的因素可能还包括红细胞生成素的升高、免疫系统功能紊乱等。TC 可发生于任何年龄, 但以10-20岁为多,女性多于男性, 家族性发病者占33%-50%。TC 多见于儿童和青少年,无明显的性别差异。目前病例报道多集中于黑色人种,在非洲和新几内亚多发,欧洲和北美洲较少。大部分患者健康情况良好,钙沉着部位可出现肿胀、疼痛、被覆皮肤溃疡、瘘管形成或局部有白垩样物质流出。 关于本病的发病机制, 目前认为, 病灶处存在大量壁菲薄、通透性高的毛细血管, 血液往复循环于其中时携来丰富的钙及磷酸根离子; 免疫组化显示病变钙化灶边缘的组织细胞样细胞源自中胚叶, 为前骨母细胞, 其主要产生碱性磷酸酶而不产生有机的骨基质。因而在局部促成大量钙质沉着, 但无骨化改变。 Slavin等按照临床表现将TC 患者分为3 种类型:(1)患者在20岁前出现多发性病变。部分患者血磷酸盐轻至中度升高,还可出现1,25 二羟基维生素D3 升高。病变多位于大关节附近,表现为皮下缓慢性生长、质地坚韧的钙化性肿块,常与其下的筋膜、肌肉或肌腱紧密相连,但不累及骨和关节。此型可认为是先天性钙代谢异常,按常染色体显性遗传,有家族发生的倾向。(2)患者发病年龄范围很大,无高磷酸盐血症,但是血清钙盐可升高。(3)病变为继发于肾功能衰竭、各型结缔组织病、进行性骨干发育异常或唐氏综合征等疾病,患者常伴有高钙血症或高磷酸血症。 病理上将TC 分为活动期和静止期, 呈多囊性或实性, 前者囊壁及间隔内衬肉芽组织, 后者仅有纤维组织和胶原纤维构成。T C 的病理特点为成纤维组织和胶原纤维组成的包膜内, 填充乳白色石灰样糊状钙化沉积物及淡黄色乳糜状液体, 囊内见大小不等的钙化灶, 囊壁可见上皮细胞和多核巨细胞。 X线检查是诊断TC的基本方法, 其表现为关节旁关节伸侧软组织中, 呈大小不一的钙化结节集结而成的分叶状团块, 呈“卵石样”,“桑葚状”,范围较广者可呈“流注状”;病变一般不累及邻近关节或骨骼。CT 与X 线平片表现一致, 但CT 对病变部位、形态及范围的显示更为全面, 能清楚显示病变与邻近关节及骨骼的关系。MRI具有多参数、多序列、多方位成像的功能, 可根据信号判断组织成分; 由于肿瘤主要由纤维包膜包裹的钙化沉积物及淡黄色乳糜状液体组成, 内有纤维间隔,因此T1WI 肿瘤呈不均匀低信号, T2WI 呈不均匀高信号; 肿瘤包膜呈长T1 、长T2信号。MRI对观察肿瘤边缘及肿瘤与关节、骨骼的关系价值大, 能多方位显示病变不累及关节或骨骼, 对诊断有较大帮助。 肿瘤样钙质沉着症是由于多部位的羟磷灰石结晶组成的以关节周围钙化的软组织肿块为特征性的疾病、多数与磷酸过高所致的异常磷酸盐代谢有关,通常发生于肩髋等大关节。特发性肿瘤样钙质沉着症有家庭倾向,多见于肾病患者,继发性钙质沉着症的鉴别诊断,包括骨肉瘤、软骨肉瘤及滑膜肉瘤的软组织钙化和骨化,也包括良性软组织肿块中的营养不良和代谢所致的钙化或骨化,也可伴随临近的骨侵蚀,但硬皮病易累及手部,而肿瘤样钙质沉着症的特征性X线表现,即在肿块中发现不规则分叶状肿块被纤维分隔,形如鹅卵石或“鸡笼”样表现。但其他疾病,如硬皮病的肿块亦呈分叶状,导致了肿瘤样钙质沉着症与其他疾病鉴别上的困难。 本病的治疗方法是彻底切除病灶, 特别是彻底刮除位于松质骨内的钙化物质, 以防术后
尿钙的临床意义
一、尿钙是什么 成人体内钙总量约为400~800g,约99%的钙分布于骨骼和牙齿。人体每天有约80%的钙经肠道排出,20%的钙经肾由尿液排出。 每日由肾小球滤出约10g钙,其中的一半在近曲小管被重吸收,其余的在髓襟、远曲小管及集合管中被重吸收,尿中排出钙量只占滤过量的1.5%(约150mg)。机体钙、磷代谢受甲状旁腺素(PTH)、降钙素(CT)及活性维生素D3调节,并且作用于骨骼、肠道与肾小管。 尿钙检测包括半定量(试纸条法)和定量(如邻甲酚酞络合酮比色法)。半定量法一般参考范围在1.0~10.0mmol/L范围,定量(邻甲酚酞络合酮比色法)的参考范围为2.5~7.5mmol/24h。 高钙膳食、服用维生素D3、使用利尿剂或长期服用肾上腺皮质激素等,尿钙含量可能会增高。如用随机尿检测,一般配合肌酐一起检测,在计算时用肌酐值矫正。 二、尿钙异常的原因是什么 尿钙增高可见于: 生理性包括高钙膳食、维生素D3摄入过多等; 病理性包括甲状旁腺功能亢进症、特发性高尿钙症、溶解性骨癌及肉瘤转移、Paget病、结节病、骨质疏松症、肢端肥大症及肾小管损伤等。 尿钙降低可见于:
生理性如低钙膳食等; 病理性减少包括甲状旁腺功能减退症、维生素D缺乏症、佝偻病、软骨病、手足抽搐症、慢性肾衰竭、尿毒症等。 三、尿钙增多的常见疾病 甲状旁腺功能亢进症 原发性甲状旁腺功能亢进症(简称原发甲旁亢)是由于甲状旁腺肿瘤(腺瘤或癌)分泌过多的甲状旁腺激素(PTH)所引起的一种疾病。 其临床特点为高血钙、高尿钙、低血磷、泌尿系统结石和骨骼改变。发病率约0.1%,发病年龄以30~70岁居多,儿童少见;在大于50岁的人群中,女性与男性之比约为3:1。 临床表现 一般进展缓慢。轻者仅表现为肌无力、反应迟钝、食欲减退,后期可有抑郁、感觉异常、近端肌无力、肌萎缩等。重者可有消化道溃疡病样症状,以及多尿、多饮、脱水及体重下降。 ①肾脏症状:由于血钙过高致尿钙排出增多,又因尿磷排泄增加,故患者常有烦渴、多饮和低渗尿等。尿路结石的发生率较高,以多发、反复发作和活动性小结石为特点。尿结石和肾实质钙盐沉着对本病具有诊断意义,可致肾绞痛、血尿、尿路感染和肾功能不全。 ②骨骼症状:有广泛性骨关节疼痛伴明显压痛。绝大多数患者骨密度减低,重者有骨骼畸形。约50%以上的患者有自发性病理性骨折和纤维囊性骨炎。
钙沉积诊断详述
钙沉积诊断详述 *导读:钙沉积症状的临床表现和初步诊断?如何缓解和预防? 1.焦磷酸钙沉积病的诊断主要依靠①滑液或组织(主要是关节囊、腱鞘的活检)中焦磷酸钙晶体存在的直接证据;②关节或软组织的X线表现其他的一些临床或实验室检查多用于除外其他的 疾病或是诊断患者同时是否伴有其他关节疾患而焦磷酸钙沉积 病的诊断一旦成立,最好进一步探究其病因,特别是追溯该病是否继发于一些遗传代谢病的可能。 2.焦磷酸钙沉积病的诊断标准 Ⅰ.通过红外光谱或X线衍射的方法在关节滑液或病理标本中发 现明确的焦磷酸钙晶体。 Ⅱ(a)在相差偏振光显微镜视野下见到标本中有弱正性双折射光 或无折射光单斜晶或三斜晶的存在。 Ⅱ(b)在X平片上发现纤维软骨或透明软骨有典型的钙质沉着。 Ⅲ(a)临床上有急性关节炎的表现,特别是当累及膝关节或其他 一些大关节时 Ⅲ(b)临床上主要表现为慢性关节炎,可以呈现急性发作,膝髋、腕、肘、肩或掌指间关节更易累及。 根据标准Ⅰ或标准Ⅱ(a)十Ⅱ(b)可诊断焦磷酸钙沉积病。 根据标准Ⅱ(a)或Ⅱ(b)可诊断可能的焦磷酸钙沉积病。 根据标准Ⅲ(a)或Ⅲ(b)临床上仅提示有焦磷酸钙沉积病存在的
可能。 钙化灶:是指用B超或ct图像上测到的某器官的出现类似结石一样的强回声或高密度影像的钙质沉淀。常见有肝钙化灶、前列腺钙化灶、肾钙化灶等。 钙离子内流:钙离子通过活化钙调素能调节众多的细胞生物学过程;钙调素是人体的一种重要的钙结合蛋白,作为钙离子作用的受体,是协助钙离子完成多种生理机能的媒介。钙离子与癫痫发作的关系已经明确,钙离子细胞内流是癫痫发病的基本条件。 钙化:病理学上指局部组织中的钙盐沉积,常见于骨骼成长的早期阶段,亦见于某些病理情况下(如结核病干酪样坏死病灶中的 钙化)。 血中钙离子过高:血钙的正常值为100毫升血液中含钙9—11毫克,即每升血中2.2—2.7毫摩尔浓度。血钙的正常波动幅度较小,主要是钙对维持人体多种生理功能极为重要。通过血钙离子的检测能够判断多种疾病的可能性。例如:甲状旁腺机能亢进时,血离子钙高于正常范围。 维D摄入过量使钙与磷结合导致钙沉积,多喝白开水,多食黑木耳,少吃鱼肝油,睡前少喝牛奶,少吃豆制品等。 *结语:以上就是对于钙沉积的诊断,钙沉积怎么处理的相关内容介绍,更多有关钙沉积方面的知识,请继续关注或者站内搜索了解更多。
阴囊特发性皮肤钙沉着症6例
阴囊特发性皮肤钙沉着症6例 阴囊特发性皮肤钙质沉着症是一种原因不明的少见病[1],2009年3~6月在本院保健科婚检中心发现6例病例。 1 临床资料 患者均为男性,年龄22~28岁,病程为半年至6年,结节数为8~26个,大小为0.5~1.5cm。患者均为婚检对象,均无明显诱因,无意中发现的,无不适。先在阴囊皮肤出现绿豆大的丘疹,后丘疹逐渐增大变硬,且数量增多,有的双侧阴囊皮肤同时出现,有的先单侧然后逐渐波及另一侧,丘疹逐渐增大后变硬,顶部变白可自行破溃,可挤出白色豆渣样物质,随着病程的过展,皮损逐渐向周围扩散,但仅局限于阴囊皮肤,因无不适均未予治疗。但其中有一患者阴囊皮损破溃自己用灭菌磺胺颗粒撒在患处已结痂。患者即往体健否认全身性疾病史,家族无慢性疾病及类似疾病,否认遗传病,代谢性疾病,父母也不是近亲结婚。体检:生命体征及各系统均正常。皮肤科情况:阴囊皮肤两侧均可见大小不等、形如黄豆、绿豆的结节、质硬、界清、无活动,小的皮损表面颜色正常、大的皮损顶部呈现乳白色,挤破或刺破可挤出乳白色的乳酷状物。 2 讨论 皮肤钙沉着症按病因分为:特发性皮肤钙沉着症和转移性皮肤钙沉着症。阴囊特发性皮肤钙沉着症是特殊临床类型[2]。 皮肤钙质沉着症是指不溶于钙盐沉积于皮肤组织。临床表现为坚硬的丘疹、结节或肿块,沉积的钙盐主要是无定形的磷酸钙,少量碳酸钙和极少的羚磷灰石。皮肤钙质沉着症分成原发性及继发性两大类。前者原因不明,有学者认为系上皮囊肿炎症后钙化形成。后者主要因为组织损伤后引起[1]。组织损伤后引起的钙质沉着可能与损伤后释放碱性磷酸酯或脂肪坏死释放游离脂肪酸与钙离子结合产生钙有关[3]。还有学者认为致病因素包括:广泛组织损伤,如皮肌炎、系统性硬化症等;局部组织损伤,如炎症、外伤;钙磷代谢异常[4]。本病可通过手术切除或用微波、电离子等物理治疗。由于病例少,对本病的研究还少,认识不深,有待于今后工作进一步研究。
尿毒症钙化防御【最新】
尿毒症患者出现的钙化防御综合征,是由于伴有继发性甲状旁腺功能亢进引起的皮下组织和小动脉转移性钙化,从而导致皮肤缺血性溃疡。 尿毒症患者发生钙化防御是Bryant和White于1898年首先在Guy’s Hospital Reports报道的。Selye在1962年把钙化防御这一名词创新性定义为:不同器官的急性局部钙化并等同于过敏反应。其实这并不合适,因为和IgE介导的I型变态反应没有联系。他在鼠体内预先用大剂量维生素D类似物或甲状旁腺激素局部注射作为刺激物诱导广泛的软组织钙化。这些实验条件和临床环境不能相比拟,因缺乏尿毒症和小动脉中层钙化和内膜增生引起的缺血性坏死模型。因此,这一名词的适用性值得怀疑。然而,钙质沉着症不能区别坏死组织中继发性钙化和动脉钙化引起的缺血性组织坏死。 Hafner等在1995年认为这种现象是尿毒症的小动脉疾病,其特点是继于皮下组织和肢端动脉的中层钙化及内膜增生所致的皮肤坏死和肢端坏疽。 【发病机制】 目前仍不是十分清楚。慢性肾功能衰竭,甲状旁腺功能亢进,高磷饮食是引起高钙-磷乘积的易感因素(正常值:4.2-5.6mmol/l),高钙磷乘积导致钙磷结晶沉积。这导致小到中等大小的动脉和微动脉的中和内弹力层弥漫钙化和内膜增生,稀有动脉阻塞引起组织坏死。慢性肾功能衰竭血液透析患者出现皮肤钙质沉着症预示血管钙化伴有
缺血性皮肤松解。其他触发因素包括静脉内注射右旋糖苷铁和白蛋白,低白蛋白血症,糖皮质激素和免疫抑制剂的应用,创伤,肥胖病人皮下注射,蛋白C和S缺乏等都可以引起高凝状态并继发血栓形成。肥胖症被认为是危险因素是因为大量脂肪组织沉积可能引起局部血流减少有关。糖尿病引起的慢性肾功能衰竭尤其容易发生肢端坏死,部分是由于广泛的血管钙化。不同器官的间质和结缔组织中钙-磷晶体的沉积被认为是系统性钙质沉着症或转移性钙化。尽管血透患者和接受肾移植的患者血管钙化的发生率很高,但是伴有血管钙化的组织坏死很少。人类钙化防御的病例从网上文献资料来看,PubMed目录中1999.7.1是285例,到2000.7.4增加到377例。这个数据也包括61例透析患者眼钙化防御和30例器官-软骨-骨钙化防御,这一状态很难和没有钙磷代谢异常而只是钙盐沉积在气管粘膜引起的钙化防御区别。大量动物研究和无此综合征的患者也被纳入研究。 【临床特点】 损害开始是疼痛,对称性的,紫罗兰色脱色,并进展位点装分界,没有愈合的溃疡变为坏死和坏疽。近心端的肩膀和躯干,臀部,大腿的损害,预后不好,主要是由于大块坏死和感染组织。远端损害包括小腿,前臂和肢端(手,手指,足,足趾)和外生殖器。 复习文献没有发现这种疾病的起病年龄和结果之间的联系。起病年龄从6个月到83岁,平均年龄48+-16岁。女性居多(61%),由于
可治性罕见病—低碱性磷酸酯酶血症
可治性罕见病—低碱性磷酸酯酶血症 一、疾病概述 低碱性磷酸酯酶血症( hypophosphatasia,HPP)是一种罕见的以骨和/或牙齿矿化障碍,伴血清碱性磷酸酶( alkaline phosphatase,ALP)活性降低为特征的遗传性骨代谢疾病。其临床表现轻重不等,轻者可仅表现为成年后下肢病理性骨折,严重者可致无矿化骨的死胎[1]。临床上根据诊断年龄和表型严重程度可分为6种亚型,分别为围生期(重型)、围产期(轻性)、婴儿型、儿童(少年)型、成人型、牙型。国内患病率尚不详,欧洲重型HPP的患病率约1: 300 000,其他型HPP患病率约1:6 300[2]。HPP是由编码组织非特异性碱性磷酸酶(tissue-nonspecific alkaline phosphatase,TNSALP)的ALPL基因变异所引起。正常TNSALP可水解焦磷酸盐为无机磷,与钙结合形成羟基磷灰石,促进骨矿化。ALPL基因变异引起TNSALP折叠、组装、调节或转运发生异常,酶活性下降,TNSALP底物如焦磷酸盐、吡哆醛等堆积,引起骨骼矿化障碍,而大量血钙无法以磷酸钙形式在骨内沉积,导致血钙升高[3]。ALPL基因位于1p36.12,可以常染色体隐性遗传方式或常染色体显性遗传方式致病。 二、临床特征 不同分型的HPP临床表现不同。 1.围生期(重型) HPP最严重的一型,通常通过产前超声检查而发现,严重者因骨矿化障碍出现死胎,幸存至出生的新生儿因胸部畸形如胸腔小、连枷胸等导致肺功能不全,常于出生数天因呼吸系统并发症而死亡[4]。四肢短呈弓形,可伴高钙血症、癫痫发作或呼吸暂停。 2.围产期(良性) 产前超声检查显示长骨短呈弓形,伴正常或轻度下降的骨矿化。这类患儿出生后骨骼异常可缓慢改善,预后良好[5]。 3.婴儿型 出生时正常,出生后6月龄之内可出现佝偻病表现。临床严重程度取决于肺功能不全的程度。病死率较高,50%患儿死于矿化不全的肋骨所致呼吸系统并发
特发性家族性脑血管亚铁钙沉着症
MMIBIMONTHLYVol13No.4Aug2004 294。 2KingTD,MillsNL.Nonoperativeelosureofartrialseptaldefect.Surgery,1974;75:383—388. 3李志中,韩玲,金梅等。应用Amplatzer封堵器治疗动脉导管未闭。中华心血管病杂志,2000,5:371。373。 4张戈军,戴汝平,刘延龄等。经导管置入CardioSEAL封堵器治疗房问隔缺损及其疗效评价。中华心血管病杂志,2001,5:327~330。 5戴汝平,刘延龄,张戈军等。应用Amplatzer封堵器治疗房间隔缺损疗效评价。中华心血管病杂志,2000,28:87~92。6张戈军,戴汝平,刘延玲等。房间隔缺损封堵术后心功能的变化。中华心血管病杂志,2001,29(3):163~166。 7徐立,戴汝平,刘延龄等。Amplatzer法房间隔缺损封堵术一经食道超声心动图测量值选择封堵器的可能性。中华心血管病杂志,2001,29:297—299。 8张玉奇,陈树宝,孙琨等。双平而食管超声心动图在Am—platzer封堵器经心导管关闭小儿房间隔缺损中的价值。中华超声影像学杂志,2001,10(11):651—653。 9朱振辉,刘延玲,戴汝平等。经胸超声心动图监测引导经导管房间隔缺损封堵术的临床研究。中华超声影像学杂志,2001,10(8):455—458。 特发性家族性脑血管亚铁钙沉着症 武警陕西总队医院放射科(西安710054)陈毓秀徐杰牛灵芝 特发性家族性脑血管亚铁钙沉着症,又称Fain(法氏病)或称家族性基底节钙化,它是以双侧基底节、丘脑、小脑齿状核及皮质下中枢对称性钙质沉着为主要病理学特征的疾病,临床资料报道较少,笔者在工作中遇到1例,现报道如下:患者男,60岁,以高血压心脏病收住院。既往有头痴病史,为慢性钝痫。查体:神清合作,一般状况好,神经系统及各脏器检查未见明显异常。实验室检查:血钙2.6mmol/L,血磷1.5mmol/L,头颅CT平扫;双侧丘脑、壳核、苍白球、尾状核可见大量斑片状钙化灶,CT值220Hu一700Hu(图1),双侧侧脑室、三脑室、中脑导水管及四脑室均无扩大、变形,中线结构无移位(图2),Cr诊断:家族性基底节钙化。 图1基底节区(双侧)J。泛刈称肚钙化灶。图2脑室系统形态正常。 讨论:本病较为少见,病因不明,有家族倾向,多为常染色体显性或隐性遗传,病理表现病变区广泛对称的终末小动脉和静脉周围的钙质沉着。临床表现为进行性精神障碍,智力低下、痴呆、语言障碍,严重生长障碍和癫痫。但笔者在临床工作中遇到两例,均无明显上述症状,体格发育良好,智力发育正常,均从事正常日常工作,生活自理,仅仅表现不规则性慢性头痛,是否可以考虑为这类患者存在有血管发育异常,这与吴恩惠教授的有关报道相吻合,对于此类患者是否进一步行脑血管造影,【:包有待于进一步的观查和探讨。 本病要注意与甲状旁腺机能低下、结节性硬化相鉴别;前者有实验室检查,而后者在Cq"影像上呈不对称性高密度 影。 万方数据
慢性肾衰的解释
慢性肾衰 慢性肾衰是由各种慢性肾脏疾病引起的进行性、严重的代谢紊乱及其他损害所组成的一组症候群。慢性肾衰是一缓慢的进行性过程,也是一个不可逆的过程,但并非不治之症。随着科学的发展(如透析、肾移植等),使许多病人存活更长时间,有些病人可长期存活。常见病因为肾小球肾炎、间质性肾炎、高血压、糖尿病及梗阻性肾病。此外,多囊肾、遗传性肾脏疾病、狼疮性肾炎、镰状细胞病等也较常见。少见病因为急性肾功能衰竭未恢复、多发性骨髓瘤性肾病、肾淀粉样变、麻风、肾结核、结节性多动脉炎、韦格内氏肉牙肿、肾钙质沉着症等。 慢性肾功能衰竭(简称慢性肾衰)又称慢性肾功能不全,是指各种原因造成的慢性进行性肾实质损害,致使肾脏明显萎缩,不能维持其基本功能,临床出现以代谢产物潴留,水、电解质、酸碱平衡失调,全身各系统受累为主要表现的临床综合征,也称为尿毒症。 衰 慢性肾衰是所有进展性肾疾病的最终结局,因此慢性肾衰的病因多种多样,其常见的病因主要有: ①慢性肾小球肾炎,如IgA肾病、膜增殖性肾小球肾炎;局灶节段性硬化性肾小球肾炎和系膜增殖性肾小球肾炎等;
②代谢异常所致的肾脏损害,如糖尿病肾病、痛风性肾病及淀粉样变性肾病等; ③血管性肾病变,如高血压病、肾血管性高血压、肾小动脉硬化症等; ④遗传性肾病,如多囊肾、Alport综合征等; ⑤感染性肾病,如慢性肾盂肾炎、肾结核等; ⑥全身系统性疾病,如狼疮性肾炎、血管炎肾脏损害、多发性骨髓瘤等; ⑦中毒性肾病,如镇痛剂性肾病、重金属中毒性肾病等; ⑧梗阻性肾病,如输尿管梗阻;反流性肾病、尿路结石等等。另外,大约有6%—9%的患者病因难以确定。据国外的研究表明,在慢性肾衰行血液透析的患者中,占第一位的是糖尿病肾病,约为27.7%,第二位的是高血压肾损害,约占22.7%,慢性肾小球肾炎占第三位,约为21.2%,多囊肾为3.9%,其他各种病因共占24.5%。中国尚没有慢性肾衰病因大规模调查的资料,从临床经验来看,中国慢性肾衰的病因仍以慢性肾小球肾炎为主,其次是肾小管间质性疾病。最近几年由于生活方式的改变,由此引起的疾病在中国的患病率大量增加,糖尿病和高血压所致慢性肾衰的患者数量也大幅增多,这一动向应该引起临床医师的注意。[1]
维生素D缺乏症
儿童维生素D不足与缺乏 维生素D是一种必需营养素,对维持钙稳态和骨骼健康具有重要作用。严重维生素D缺乏会导致婴儿和儿童发生佝偻病和/或低钙血症,后者可引起手足搐搦或抽搐。这些疾病在营养不良儿童群体和慢性疾病儿童群体中的发生率最高。即使没有发生佝偻病,维生素D水平长期较低也与骨密度低和其他骨骼健康指标降低相关。 一、维生素D的代谢与形式 维生素D作为一种激素前体,由皮肤暴露于紫外辐射后合成。在没有食品强化或补充剂的情况下,膳食来源的维生素D不到10%。维生素D随后在肝脏和肾脏中被转化成其代谢活性形式。 ●胆钙化醇,或称维生素D3,这是动物制品和某些维生素D补充剂中的维 生素D形式。 ●麦角钙化醇,或称维生素D2,由植物中的麦角固醇暴露于辐射而生成。 它是膳食植物来源和大多数维生素D补充剂中的维生素D形式。 ●维生素D与维生素D结合蛋白结合后,被转运至肝脏;在肝脏中,维生 素D通过25羟化作用转变为25羟维生素D[25(OH)D],这是维生素D的贮存形式,也称为骨化二醇。 ●在肾脏中,25(OH)D经过1-α羟化作用形成1,25(OH)2维生素 D[1,25(OH)2D],此为维生素D的活性形式,也称为骨化三醇。
●维生素D充足:25(OH)D大于等于20ng/mL(50nmol/L) ●维生素D不足:25(OH)D介于15-20ng/mL(37.5-50nmol/L) ●维生素D缺乏:25(OH)D小于等于15ng/mL(37.5nmol/L) 2、维生素D缺乏的原因 a合成减少 阳光暴露(特别是UVB)是皮肤合成维生素D必不可少的因素,而肤色深的儿童因为黑色素起着自然防晒霜的作用,故而阳光暴露减少。 居住地纬度和季节也是皮肤维生素D合成的重要决定因素。冬季在高于纬度40°的地区,到达地球表面的UVB辐射甚微。因此,虽然维生素D缺乏在夏季末相对少见,但在冬季末则很常见。
Fahr综合征
Fahr综合征 又名:Fahr综合征;大脑钙质沉着伴晚发性脑病;大脑钙质沉着;基底节钙化;对称性大脑钙化综合征。特发性两侧对称性大脑基底节钙化症;特发性家族性脑血管亚铁钙沉着;家族性基底节钙化。 对称性大脑钙化综合征于1930年由Fahr所描述。国内有少数报道。 【病因】未明。一般认为是假-假性甲状旁腺机能减退(Albright遗传性骨营养不良症)的一种异常类型,是常染色体隐性遗传,多伴有因甲状旁腺功能缺陷而致之钙-磷代谢改变,钙、磷离子与含大量蛋白质的有机基质相结合。 发病机制:是肾脏对甲状旁腺(PTH)正常下的反应,血清钙磷正常,钙化起始于基底节和齿状核,钙化的过程与生理性钙化相似,随疾病的进展、钙沉着的程度和范围也增加,可能因血管壁钙化所致的血管闭塞而造成脑萎缩,钙质沉着不一定伴神经系统症状。 【临床表现】多数病例有钙质沉着,但无神经系统症状,仅于脑部X线检查时发现,一般可于45~60岁出现症状。主要为精神衰退、锥体外系统性运动缺陷、小脑性共济失调、震颤症状群及构音障碍、情感迟钝、记忆减退,有的表现焦虑、抑郁伴偏执、妄想、精神病样症状。颅部X线:双侧基底节对称性钙化,脑CT检查除双侧基底节、小脑齿状核和脑沟处可见钙化外,还可发现大脑轻度萎缩和脑室扩大。 【诊断】自幼开始出现慢性进行性智能衰退和锥体外系受损症状,结合颅部X线的表现以及脑CT的可靠证据确诊。本病应与下列疾病鉴别:①特发性甲状旁腺机能减退:是由于甲状旁腺激素不足,造成低血钙,高血磷及Ellsworth-Howard试验(甲状旁腺激素磷利尿试验)阳性。②假性甲状旁腺功能减退:系因骨骼和肾小管对甲状旁腺激素有对抗所致。有低血钙、高血磷和Ellsworth-Howard试验阴性,血清甲状旁腺素浓度高于正常。③假-假性甲状旁腺机能减退:血清钙、磷浓度正常,骨骼和肾小管对甲状旁腺激素的反应也正常,即Ellsworth-Howard试验正常。假性和假-假性甲状旁腺功能减退之患者都具有特征性躯体表现:身材短小,圆脸,掌跖骨短,牙齿异常,白内障和软骨组织钙化等;而Fahr综合征仅有大脑钙质沉着及神经精神症状,并无上述躯体特征,可作区别之条件。 【治疗】无特殊疗法,予对症处理。
特发性钙质沉着症1例
特发性钙质沉着症1例 发表时间:2011-12-07T11:32:26.447Z 来源:《中国医药卫生》2011年第9期作者:周晓倩 [导读] 经脱钙后制片,镜检:皮下组织及真皮内见片、灶颗粒状钙盐沉着,其周围异物性肉芽反应不明显。 周晓倩 内蒙古大兴安岭农管局中心医院(内蒙古大兴安岭165456) [中图分类号]R758.2[文献标识码]A[文章编号]1810 5734(2011)9-0115-01 1病例摘要 患者,男,64岁。30年前,自觉阴囊皮肤下方生长一肿物,约豆粒大小,无明显不适感,当时未予治疗。后肿物于阴囊皮肤下方逐渐增大,并逐渐增多。入院前10天,自觉肿物疼痛,伴瘙痒,为求明确诊治来院。无发热及冶游史,病程中饮食、睡眠及二便均正常,入院查体:T36·5℃,神志清晰,语言流利。皮肤无丘疹及瘀斑。心、肺无异常。肝、脾无肿大。阴囊皮肤可见多发、散在肿物,皮肤无溃疡,无脓性分泌物。各包块生长于阴囊皮肤表面,高于皮表,肿物呈纵行排列,左右阴囊均有,质略硬,不活动,轻压痛。脊柱、四肢无畸形,无病理性骨折。双肾无扣击痛。关节无红肿,活动自如,肌张力正常。 实验室检查:血、尿常规正常,肝、肾功能正常,心电图、X线胸部正侧位片均正常,电解质、血沉等基本正常,肝、胆、脾、胰及肾未见明显异常。 病理检查:术后标本巨检,切除灰褐色表皮组织2块,表面带毛发,分别为9.5cm×3cm和6.2cm×2cm。皮表向外隆起多个(共13个)结节性肿物,直径0.5~2cm不等,切面灰白,实性,其间可见点、灶状石灰样物,质硬,有砂砾感。经脱钙后制片,镜检:皮下组织及真皮内见片、灶颗粒状钙盐沉着,其周围异物性肉芽反应不明显。病理诊断:特发性钙质沉着症。 2讨论 阴囊皮肤可发生原因不明的钙盐沉着,很可能是皮脂腺囊肿破裂钙盐沉积的结果,常发生于年轻人,病变可单发,但常多发,触之为硬结。若破溃可有白垩样物流出。病变广泛者,阴囊皮肤呈硬壳状。病理检查于真皮及皮下组织内见颗粒状钙盐沉着,呈灶性或大的团块,沉着物周围见异物性肉芽反应。钙盐在HE下为浅蓝色无定形均质状或颗粒状,Von ko-ssa特染呈黑色。 血钙代谢障碍,甲状旁腺机能亢进及一些胶原结缔组织病也可以引起皮下钙盐沉着,但往往是系统性的,常累及多部位,多器官,不只限于阴囊。此外血钙代谢障碍以及甲状旁腺机能亢进常表现为高血钙症候群,胶原结缔组织病则常有不明原因的发热,不同程度的皮肤、关节、内脏的损害及血沉增高等共同特征。 特发性钙质沉着症是阴囊瘤样病变中比较特殊的一型,此例临床不多见。诊断时要详细询问病史,强化各项相关检查,以排除可以引起皮下钙盐沉着的其他疾病。〖FL)〗
Fahr综合征
Fahr综合征 又名:Fahr综合征;大脑钙质沉着伴晚发性脑病;大脑钙质沉着;基底节钙化;对称性大脑钙化综合征。特发性两侧对称性大脑基底节钙化症;特发性家族性脑血管亚铁钙沉着;家族性基底节钙化。?对称性大脑钙化综合征于1930年由Fahr所描述。国内有少数报道。?【病因】未明。一般认为是假-假性甲状旁腺机能减退(Albright遗传性骨营养不良症)的一种异常类型,是常染色体隐性遗传,多伴有因甲状旁腺功能缺陷而致之钙-磷代谢改变,钙、磷离子与含大量蛋白质的有机基质相结合。?发病机制:是肾脏对甲状旁腺(PTH)正常下的反应,血清钙磷正常,钙化起始于基底节和齿状核,钙化的过程与生理性钙化相似,随疾病的进展、钙沉着的程度和范围也增加,可能因血管壁钙化所致的血管闭塞而造成脑萎缩,钙质沉着不一定伴神经系统症状。 【临床表现】多数病例有钙质沉着,但无神经系统症状,仅于脑部X线检查时发现,一般可于45~60岁出现症状。主要为精神衰退、锥体外系统性运动缺陷、小脑性共济失调、震颤症状群及构音障碍、情感迟钝、记忆减退,有的表现焦虑、抑郁伴偏执、妄想、精神病样症状。颅部X线:双侧基底节对称性钙化,脑CT检查除双侧基底节、小脑齿状核和脑沟处可见钙化外,还可发现大脑轻度萎缩和脑室扩大。 【诊断】自幼开始出现慢性进行性智能衰退和锥体外系受损症状,结合颅部X线的表现以及脑CT的可靠证据确诊。本病应与下列疾病鉴别:①特发性甲状旁腺机能减退:是由于甲状旁腺激素不足,造成低血钙,高血磷及Ellsworth-Howard试验(甲状旁腺激素磷利尿试验)阳性。②假性甲状旁腺功能减退:系因骨骼和肾小管对甲状旁腺激素有对抗所致。有低血钙、高血磷和Ellsworth-Howa rd试验阴性,血清甲状旁腺素浓度高于正常。③假-假性甲状旁腺机能减退:血清钙、磷浓度正常,骨骼和肾小管对甲状旁腺激素的反应也正常,即Ellsworth-Howard试验正常。假性和假-假性甲状旁腺功能减退之患者都具有特征性躯体表现:身材短小,圆脸,掌跖骨短,牙齿异常,白内障和软骨组织钙化等;而Fahr综合征仅有大脑钙质沉着及神经精神症状,并无上述躯体特征,可作区别之条件。?【治疗】无特殊疗法,予对症处理。