GST融合蛋白构建、表达与纯化
蛋白质表达:带GST标签蛋白的纯化

操作前的考虑
• BugBuster蛋白抽提试剂,以及Benzonase®核酸酶、rLysozyme™溶菌酶等能极大地方便可溶蛋白 及包涵体的制备,从而完全替代机械法操作,尽可能地保全蛋白。
• BugBuster,Benzonase核酸酶,rLysozyme溶菌酶及GST•Bind系统均可与蛋白酶抑制剂兼容。 • GST•Bind纯化系统可以与2-巯基乙醇,二硫苏糖醇以及EDTA等兼容。 • 本手册中的操作均可按实际表达水平和目的蛋白的量适当加以调整,特别是关于“柱层析”部分中的
默克生命科学服务热线:400 820 8872 bioteam@merc这样抽提得到的可溶蛋白溶液可以直接上 样于Novagen的纯化树脂(以及其它很多类似纯化系统)。蛋白溶液 在冰上可以短时存放(2-3小时),也可在-20℃长时间存放直至下步 分析。蛋白抽提液应该根据目的蛋白的活性要求的温度存放,有些蛋 白经冻融会失活。
储存
要求4℃存放的包括:10X GST Bind/Wash Buffer;10X Glutathione Reconstitution Buffer; Glutathione,Reduced,Free Acid;GST•Bind树脂(树脂千万不能冻) 要求-20℃存放的包括:Benzonase核酸酶 常温存放的包括:BugBuster,色谱柱
GST•Bind™ 缓冲液试剂盒
GST•Bind缓冲液试剂盒包括了从GST•Bind树脂和GST•Mag™磁性琼脂糖树脂纯化目的蛋白所需的结 合,冲洗和洗脱所需的缓冲液。试剂盒所含成分足够用于10个2.5ml柱的纯化。具体包括: • 2×100ml 10×GST Bind/Wash Buffer (43mM Na2HPO4,14.7mM KH2PO4,1.37M NaCl,27mM KCl,pH7.3) • 40ml 10×Glutathione Reconstitution Buffer (500mM Tris-HCl,pH8.0) • 1g Glutathione,Reduced,Free Acid
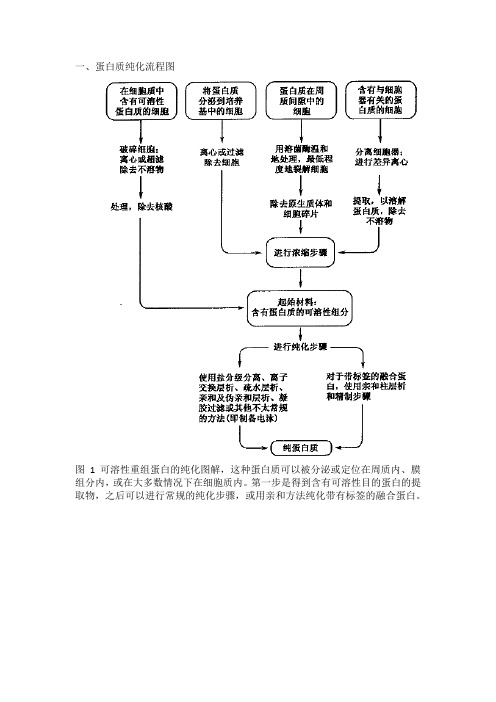
GST融合蛋白表达与纯化
70%(V/V)乙醇
适于水溶液的0.22μm膜
1、在1.5ml的微量离心管中放入1ml表达目的蛋白的大肠杆菌细胞培养物,4℃,最高转速离心2min,收集上清,将其过0.22μm的膜,收入1.5ml的微量离心管中,弃沉淀。
材料(带√的项见附录1)
用含有trp启动子和目的基因的质粒转化的大肠杆菌宿主菌株
√M9最低营养培养基加0.5%(m/V)酪蛋白水解物(Difco公司)
√1000x抗生素贮液
√400x吲哚-3-丙烯酸(IAA)
20ml无菌培养管
150ml摇瓶
37℃定轨摇床
1、从新鲜的琼脂平板上挑取含trp启动子和目的蛋白基因质粒转化的大肠杆菌宿主菌株的单一菌落,接种至含有2ml M9培养基(含0.5%的酪蛋白水解物和适量的抗生素)的20ml培养管,将培养管在定轨摇床上(200rpm)37℃培养过夜。
转化的大肠杆菌宿主菌株
√LB培养基
√1000x抗生素贮液
√20%(V/V)甘油,无菌的
√LB平板
20ml培养管
30℃或37℃定轨摇床
1、从新鲜琼脂平板上挑单菌落,接种2ml LB培养基(补加适当的抗生素;在20ml培养管中),在定轨摇床上培养过夜。对于基于pL的系统,温度为30℃,对于其他系统,温度为37℃。
辅助方案5 细胞外培养基样品的制备
通过分析培养基中的样品能够检查指导目的蛋白细胞外分泌的表达系统。如果培养物中蛋白质浓度很低(如低于100mg/L),可以加入牛血清白蛋白(BSA)作为沉淀的载体和核。
材料
表达目的蛋白的转化大肠杆菌细胞
2g/L BSA(可选;用0.22μm的膜除菌,4℃保存≤1个月)
gst蛋白纯化

GST蛋白纯化简介谷胱甘肽S-转移酶(glutathione S-transferase,GST)是一种常用的亲和标签,用于在分子生物学研究中用于蛋白质纯化和蛋白质亲和结合实验。
GST蛋白被广泛应用于蛋白质结构和功能研究、酶学研究、蛋白质互作研究等领域。
本文将介绍一种常见的方法来纯化GST蛋白,该方法主要包括以下步骤:细胞裂解、亲和层析、洗脱和纯化。
方法细胞裂解首先需要将GST蛋白表达在适当的宿主中,例如大肠杆菌。
在细胞达到适当的生长阶段后,使用合适的方法将细胞裂解,使得目标蛋白释放到溶液中。
一种常用的裂解方法是超声波裂解,通过超声波震荡将细胞破碎。
亲和层析经过细胞裂解后,将得到的细胞裂解液通过亲和层析柱。
亲和层析柱通常使用含有还原型谷胱甘肽(glutathione,GSH)结合物质的树脂,例如glutathione agarose beads。
这种树脂与GST标签结合,使得GST标签的融合蛋白能够特异性地结合于柱子上。
通过洗脱液去除非特异结合蛋白,将目标蛋白纯化。
洗脱洗脱过程是将结合于柱子上的目标蛋白从固定相洗净。
一般采用含有高浓度还原型谷胱甘肽的洗脱液,例如50 mM GSH。
洗脱液中的还原型谷胱甘肽与柱子上的结合物质竞争与GST标签结合,以此达到将GST蛋白洗脱下来。
纯化经过洗脱后,蛋白溶液中的GST蛋白含量较高。
为了进一步提高纯度,可以通过对溶液进行浓缩、去除低分子量杂质、调整溶液pH值等方法进行纯化。
常用的纯化方法包括丙酮沉淀法、离子交换柱层析法等。
注意事项•在实验过程中应严格操作,避免任何可能导致目标蛋白污染的情况发生。
•选择合适的表达宿主,不同的宿主可能会对GST蛋白的表达量和可溶性产生影响。
•在细胞裂解过程中,避免样品受到温度、剧烈振荡等因素的影响。
•注意亲和层析柱的操作方法,避免破损或污染。
•洗脱过程中注意还原型谷胱甘肽浓度和洗脱液的pH 值。
结论GST蛋白纯化是一种常见的蛋白质纯化方法,通过亲和层析技术可以实现对GST蛋白的高效纯化。
GST融合蛋白的纯化

GST融合蛋白的纯化诱导和收集菌体在一定的诱导条件下IPTG诱导蛋白的合成。
18~25℃的低温条件下培养可以使大部分蛋白融合蛋白可溶性表达,并保持较高的活性。
IPTG浓度一般为0.1~1.0mM。
5000rpm 5min离心收集菌体。
亲和层析柱的制备取存放谷胱甘肽琼脂糖的瓶子颠倒数次,使其混匀,取1.5ml混合液加入层析柱中,加10ml 20%乙醇,使琼脂糖在柱中自然沉降。
将20%乙醇流尽后,加10ml PBS清洗柱子,待管中PBS液面刚好没过凝胶时,套上滴口的套子,待用。
每100ml菌液的菌体用4ml PBS(加1%Triton-100、蛋白酶抑制剂)悬浮。
在冰水中超声波破碎细胞(1分钟/次×5次,每次间隔1分钟)。
将裂解液分装至小管,4℃10000rpm离心5分钟。
收集上清液,加DTT至终浓度为1mM。
0.45um过滤后加入亲和层析柱。
室温下使混合液自然通过层析柱,保留0.5ml过滤液做PAGE电泳检测用。
用10ml PBS洗柱子3遍,每次临近结束时收集洗涤液0.5ml测OD值。
配制10mM的还原型谷胱甘肽溶液,即洗脱液3ml(0.009g溶于3ml 50mM Tris-Cl溶液中)。
用洗脱液洗脱GST融合蛋白,每管0.5ml接收洗脱液。
测各管洗脱液蛋白浓度。
PAGE电泳检测纯度。
亲和层析柱的再生:用0.04M NaOH洗10ml×3次,用10ml PBS平衡后,加20%乙醇储存于4度。
或者按照beads使用说明书上的方法再生。
如果洗脱液中的还原型谷胱甘肽对实验有影响时,需用分子筛去除。
如果需要不带标签的蛋白,则蛋白被柱子吸附后,用蛋白酶进行切割;或者用分子筛过滤后,在筛子上进行酶切。
gst纯化蛋白步骤

gst纯化蛋白步骤GST纯化蛋白是一种常用的蛋白质纯化方法,它利用谷胱甘肽硫转移酶(Glutathione S-Transferase,GST)的亲和性,将GST标签蛋白与GST结合亲和树脂进行结合,然后通过洗脱,最终得到纯化的目标蛋白。
下面将为大家介绍GST纯化蛋白的详细步骤。
1.构建重组蛋白表达载体:首先需要在目标蛋白编码基因的N端或C端加上GST标签,通常选择GST-N和GST-C两种方式。
将GST标签与目标蛋白基因连接后,将其插入合适的表达载体中。
2.转化宿主细胞:将构建好的重组表达载体转化到适合的宿主细胞中,常用的宿主细胞有大肠杆菌和酿酒酵母等。
3.表达目标蛋白:宿主细胞在适宜的培养条件下进行培养,使其产生大量重组蛋白。
常见的培养条件包括温度、培养基的选择和添加诱导物等。
4.细胞破碎:培养得到丰富的重组蛋白的细胞后,通过机械或化学方法将细胞破碎。
常用的方法有超声波破碎、高压均质破碎、冻融法和溶菌酶法等。
5.蛋白纯化:将细胞破碎液进行离心分离,去除残余细胞碎片。
接下来,将蛋白样品加入含有GST结合亲和树脂的柱子中,通过亲和吸附,在树脂上富集GST标签蛋白。
6.洗脱纯化蛋白:使用合适的洗脱缓冲液,可以脱离与亲和树脂结合的非特异性结合蛋白,并洗脱纯化的目标蛋白。
一般使用还原性缓冲液,可将目标蛋白从GST结合亲和树脂上彻底洗脱。
7.蛋白质浓缩:通过合适的方法,如超滤、溶液浓缩装置等,使纯化的目标蛋白浓缩到较高浓度。
8.纯化蛋白的分析:对纯化的目标蛋白进行SDS-PAGE凝胶电泳分析,检测其纯度和分子量等指标。
通过上述步骤,可以得到较高纯度的GST标签蛋白。
需要注意的是,在步骤的选择和操作过程中,要根据目标蛋白的特性和所需纯度等要求进行调整,以获得更好的纯化效果。
希望本文对您进行GST纯化蛋白的实验有所帮助。
gst蛋白纯化原理

gst蛋白纯化原理GST蛋白纯化是一种常用的蛋白质纯化技术,其原理是利用谷胱甘肽-S-转移酶(Glutathione-S-transferase,GST)标签与谷胱甘肽的特异性结合来进行纯化。
GST标签可与谷胱甘肽通过二硫键共价亲和,然后通过GSH交换洗脱的原理进行蛋白纯化。
具体步骤如下:1.构建GST标签融合表达载体:将GST基因的编码序列与目标蛋白的编码序列融合,构建GST-目标蛋白融合表达载体。
这样,在细胞中表达该融合蛋白时,GST标签会紧密结合在目标蛋白的C端或N 端。
2.转染和蛋白表达:将构建好的GST-目标蛋白融合表达载体转染到合适的宿主细胞(如大肠杆菌),使其产生大量的融合蛋白。
3.细胞裂解和融合蛋白的亲和层析:收获融合蛋白的细胞,通过细胞裂解等方法破坏细胞膜,释放融合蛋白。
然后,将溶解的细胞提取物加载到含有谷胱甘肽固定在琼脂糖(或其他载体)上的亲和层析柱中。
GST标签可以特异性地与琼脂糖上的谷胱甘肽结合。
4.洗脱:通过洗脱缓冲液来去除非特异性结合的蛋白质,保留GST-目标蛋白复合物。
洗脱通常使用还原剂(如谷胱甘肽)、低pH 或其他方式进行。
5.目标蛋白的解离:将GST标签从目标蛋白上解离,得到纯化的目标蛋白。
这可以通过特定的酶切位点(如蛋白酶TEV切割位点)和相应的酶进行酶切,使GST和目标蛋白分别释放。
6.纯化分析:对纯化的目标蛋白进行分析,如SDS-PAGE凝胶电泳、Western blot等方法,确认目标蛋白的纯度和完整性。
在进行GST蛋白纯化时,对于融合表达载体的设计和构建、宿主细胞的选择、裂解条件和亲和层析条件的优化等方面都需要合理考虑,以获得高质量的纯化目标蛋白。
gst融合蛋白的分离和纯化的原理

gst融合蛋白的分离和纯化的原理
第一步,细胞裂解。
通常使用化学方法、物理方法或超声波等手段将目标蛋白所在的表达系统的细胞破碎,使蛋白从细胞溶液提取出来。
常用的细胞裂解方法有冻融法、超声波法和高压法等。
第二步,亲和层析。
利用GST融合蛋白在蛋白A/G琼脂糖或S-蛋白琼脂糖柱上与谷胱甘肽硫转移酶(GST)结合的特性,将目标蛋白与其他非特异性蛋白分离。
在这一步骤中,首先需要将裂解液与亲和层析树脂(例如:GST树脂)充分混合,使GST融合蛋白与亲和树脂结合;然后通过洗涤步骤,去除非特异性蛋白质的结合;最后,目标蛋白与亲和树脂的结合被破坏,并将其洗脱出来。
第三步,洗脱。
通过改变亲和层析树脂的pH值、离子浓度或添加特定试剂,破坏GST融合蛋白与亲和树脂的结合,实现目标蛋白从树脂上的洗脱。
常用的洗脱方法有酸洗脱、碱洗脱和还原洗脱。
第四步,纯化。
将洗脱的目标蛋白进行进一步的分离和纯化。
通常,还需要使用其他纯化方法,例如离心、电泳、层析等,进一步净化目标蛋白。
总的来说,GST融合蛋白的分离和纯化的原理是通过利用GST融合蛋白与亲和树脂之间的特异性结合,将目标蛋白与非特异性蛋白分离,然后通过改变条件破坏结合并将目标蛋白洗脱,最后进行进一步的纯化步骤,得到纯化后的目标蛋白。
这种方法具有简单、快速、高效的优点,在蛋白质分离和纯化的研究中得到广泛应用。
GST融合蛋白纯化方法

GST融合蛋白纯化方法1目的片段接入pGEX载体;2涂板,挑单克隆,摇菌至OD600≈1.0,加入IPTG(终浓度1 mM)诱导6-8 h;3收菌,每升菌液约以50 mL PBS重悬,加入1%Triton X-100(v/v),1%β-巯基乙醇(v/v),PMSF(终浓度1 mM);以下步骤均在冰上操作:4超声破碎菌体,15000 g,10min离心取上清,在上清中加入适量GST-beads,轻轻晃动令其吸附蛋白1 h;5 2000 g,3min离心弃上清;6加入至少10倍体积PBS,轻摇至beads悬浮于溶液中,2000 g,3 min离心弃上清;7重复步骤6 两次;8加入1 mL GST Elution Buffer,轻摇10 min;92000 g,3 min离心,收集上清;10重复步骤8-9至少两次;11 SDS-PAGE电泳检测蛋白纯度,Bradford法检测蛋白浓度;12将蛋白置于-20℃保存。
P.S. 大量提取前应取少量菌液,改变IPTG浓度,诱导温度,诱导时间等,以确定蛋白表达的最适条Thrombin cleavage(using thrombin produced by Amersham)1. Thrombin cleavage of eluted fusion protein bound to Sepharose* Mix 50 μl of thrombin (< 10 cleavage U/ml) solution and 950 μl of 1 x PBS for each ml of Glutathione Sepharose bed volumeAdd thrombin protease mixture to Glutathione Sepharose pelletGently shake or rotate the suspension at r.t. for 2-16 hCentrifuge the suspension at 500 x g for 5’ to pellet the beads and carefully transfer the eluted fraction to a clean tube.2. Thrombin cleavage of eluted fusion protein.Add 10 μl of thrombin solution (10 cleavage units) per mg fusion protein. If the amount of fusion protein in the eluate has not been determined, add 80 μl (80 U) of thrombin for each ml of Glutathione Sepharose bed volume from with the fusion protein was eluted.Mix gently and incubate at r.t. (22-25C) for 2-16 h.Once digestion is complete, GST can be removed by first removing glutathione by extensive dialysis (2,000 vol/ml) against 1 x PBS followed by batch purification.。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
GST表达融合蛋白载体pGEX-KG大小:5006bp,氨苄青霉素抗性(Amp r),IPTG诱导表达酶切位点:BamHI 930、SmaI 937、EcoRI 962、XbaI 966、NcoI 974、SalI 980、XhoI 985、SacI 992、HindIII 994GST分子量:构建pGEX-KG-YFG重组质粒1、分析所感兴趣的基因(your favorite gene, YFG)Primer Premier 5.0软件,分析YFG含有哪些酶切位点,注意是否与pGEX-KG载体的多克隆位点有重合2、确定合适的双酶切位点NEB网站()Double Digest Finder软件,查找最佳双酶切组合(下表)NEB双酶切图谱BamHI EcoRI NEBuffer EcoRI + BSA at 37°C. BamHI may exhibit star activity in this buffer.XbaI NEBuffer 3 + BSA at 37°C.At least one enzyme has < 100% activity in thisbuffer, so additional units of enzyme and/or longerincubation time may be necessary.NcoI NEBuffer 3 + BSA at 37°C.SalI NEBuffer 3 + BSA at 37°CXhoI NEBuffer 3 + BSA at 37°C.SmaI XbaINEBuffer 4 + BSA at 25°C withSmaI, then add XbaI and raisetemperature to 37°C.At least one enzyme has < 100% activity in thisbuffer, so additional units of enzyme and/or longerincubation time may be necessary.NcoINEBuffer 4 at 25°C with SmaI, thenadd NcoI and raise temperature to37°C.XhoINEBuffer 4 + BSA at 25°C withSmaI, then add XhoI and raisetemperature to 37°C.SacINEBuffer 4 + BSA at 25°C withSmaI, then add SacI and raisetemperature to 37°C.HindIIINEBuffer 4 at 25°C with SmaI, thenadd HindIII and raise temperature to37°C.At least one enzyme has < 100% activity in thisbuffer, so additional units of enzyme and/or longerincubation time may be necessary.EcoRI NcoI NEBuffer EcoRI at 37°C.3、设计PCR上、下游引物Primer Premier 5.0软件,设计PCR上、下游引物酶切位点外最多含6个碱基3’端不是A,最好是G或C,但是不推荐使用GC或CG结尾3’端至少保证有10个碱基完全配对得分(Rating)大于70[注意]上游引物:是否添加适当碱基,确保不打乱开放阅读框下游引物:添加终止密码子(UAA、UAG、UGA)4、引物合成及保存合成:上海生工生物工程技术服务有限公司(Email:******************,Tel:81767586);纯化方法:柱层析or聚丙烯酰胺凝胶电泳?;价格1.30/碱基保存:贮存浓度:100pmol/μl(100μM),工作浓度:10pmol/μl(10μM),-20°C保存5、PCR扩增YFG模板:质粒10ng/μl稀释少量-20°C保存引物:10pmol/μl(10μM)-20°C保存Taq酶:NEB Quick-Load Taq 2×Master Mix 扩增片段小于2.0kb反应条件(1)预变性94°C 5 min(2)变性94°C 30 s(3)退火待定30 s(4)延伸72°C 待定(5)重复2-5 25-30个循环(6)补平缺口72°C 10 min(7)暂存10°C[注意]退火温度:参考4(G+C)+2(A+T)-4(互补碱基),参考Ta Opt(Primer Premier 5.0)延伸时间:Taq酶:1kb/min循环数小于30,减少错配琼脂糖电泳检测PCR产物0.8%有效分离范围:10~0.8kb;1.0%有效分离浓度7~0.5kb50ml TAE加入5μl EB母液(5mg/ml)100V,30-45min拍照或者紫外灯下切胶回收6、构建pGEX-KG-YFG酶切:双酶切PCR产物、pGEX-KG回收:PCR产物直接回收、pGEX-KG电泳之后切胶回收连接:pGEX-KG 50ng、插入片段150ng转化铺平板:Amp r挑单克隆:Amp r(四个菌落足够了)鉴定:小提质粒酶切or菌体PCR7、转化BL21(DE3)pLysS菌株检测GST融合蛋白的表达(1)冰上融化BL21(DE3)pLysS感受态细胞(天根)(2)2 ml离心管中,加入25μl BL21+ 3μl质粒(300-500ng),混匀(质粒≤感受态1/10)(3)冰上放置30min(4)42°C,90s(5)冰上放置2-3min(6)加入300μl LB(无抗生素)(相当于菌体10倍体积的LB),37°C,250rpm,1 h (7)4000rpm,2min,弃上清,其余混匀以后涂平板(Amp r),37°C,过夜(16 h)(8)挑单克隆菌落,5ml LB(Amp r),37°C,250rpm,过夜(16 h)()稀释10倍、20倍、50倍测定OD600()选择合适的稀释倍数,5ml菌液,37°C,250 rpm,1 h,测定OD600(7)取100μl菌液加入5ml LB(Amp r)(50倍稀释),37°C,250rpm,过夜(16 h)(8)取500μl菌液加入5ml LB(Amp r)(10倍稀释)(其余菌液4°C保存),测OD600 (9)37°C,250 rpm,2 h(OD600:0.6-0.8),测OD600(10)室温放置20 min(摇床降温,30°C)(11)取100μl作为诱导前对照,冰上放置(12)菌液中加入IPTG,终浓度0.5-1 mM(13)30°C,250 rpm,2 -4h(可做时间梯度),测OD600(14)取100μl作为诱导后对照,连同诱导前菌液,12000rpm,离心2min,加入50μl 1×SDS 上样缓冲液(15)取4ml菌液,12000rpm,离心2min,收集到2ml离心管中(16)加入1ml GST裂解缓冲液重悬菌体(17)超声破碎细胞:超声20s,间隔10s,共3-5次,超声强度200-300W(冰浴)(18)超声后菌液换入一个新1.5ml离心管,4°C,12000rpm,离心5min(19)超声后上清:取25μl上清,加入25μl 2×SDS上样缓冲液(其余上清-80°C保存)(20)超声后沉淀:沉淀加入1ml GST裂解缓冲液重悬,取25μl,加入25μl 2×SDS上样缓冲液(其余沉淀-80°C保存)(21)将四个样品煮沸3min,12000rpm,离心5min(22)8-10%SDS-PAGE,30μl上样,顺序:诱导前菌体、诱导后菌体、超声后上清、超声后沉淀(23)考马斯亮蓝染色[注意](1)菌液OD值小于1即可(2)诱导时间最好做一个梯度,2-6h(3)诱导温度适当摸索,24°C、30°C(4)IPTG浓度可做梯度,1mM(5)超声条件可视实际情况改变,只要使细菌裂解充分即可,即菌液清亮不粘稠8、测序确认上海生工生物工程技术服务有限公司(Tel:81767529)测序引物:pGEX-3通用引物测序样品:过夜菌1ml;质粒(浓度大于50ng/μl,10μl)9、中提质粒(Promega)10、表达纯化GST融合蛋白(1)已经转化的菌液,或者重新转化BL21(DE3)pLysS(2)活化:取20μl菌液加入11ml LB(Amp r)(500倍稀释),37°C,250rpm,过夜(16 h)(3)取10ml菌液加入100ml LB(Amp r)(10倍稀释)(剩余菌液4°C保存)(4)37°C,250 rpm,1 h 10 min-1 h 20 min,OD600 0.6-0.8(OD600小于1.0即可)(5)取1ml菌液作为诱导前对照,冰上放置(6)加入IPTG,终浓度1 mM(7)30°C,250rpm,诱导适当时间(预实验确定)(8)取1ml菌液作为诱导后对照,连同诱导前菌液,12000rpm,离心2min,收集菌体,加入100μl 1×SDS上样缓冲液(9)其余菌液,分为两份,倒入50ml离心管,5000rpm,离心20 min,收集细胞(10)每管加入8-10ml GST裂解缓冲液重悬菌体,转移小烧杯中(无气泡)(11)超声破碎细胞:超声3s,间隔10s,40次左右,超声强度200-300W(冰浴)(12)将超声后菌液转移到50ml离心管中,4°C,13000rpm,离心20-30min(13)将上清转移到1个50ml离心管中,取出50μl,加入50μl 2×SDS上样缓冲液(其余上清-80°C保存)(14)将沉淀加入16-20ml GST裂解缓冲液(或PBS)重悬,取出50μl,加入50μl 2×SDS 上样缓冲液(其余沉淀-80°C保存)(15)将四个样品煮沸3min,12000rpm,离心5min(16)8-10%SDS-PAGE检测诱导、超声是否合适,30μl上样,顺序:诱导前菌体、诱导后菌体、超声后上清、超声后沉淀(17)考马斯亮蓝染色(18)混匀glutathione sepharose beads(4B)(mixed slurry),取200μl加入15ml离心管中(2份)(19)加入10ml PBS,混匀,3000rpm,离心3min,弃上清(20)加入超声后上清液,4°C轻柔摇荡60分钟(或过夜)(横放)(21)4°C,3000rpm,离心3min(22)10ml GST裂解缓冲液洗涤2次(冰上)(23)10ml TBS(含5mM MgCl2、1mM DTT)洗涤2次(冰上)(24)弃上清,加入1倍柱床体积的TBS(含5mM MgCl2、1mM DTT、50%甘油),混匀(25)取悬液测定蛋白浓度或SDS-PAGE(26)-20°C保存beads11、GST-Pull-down(1)10cm培养皿中,未处理的细胞或者转染的细胞(3-5μg质粒转染10cm培养皿,Cos-7、293T或者其他细胞,转染24h)(2)加入1ml 细胞裂解缓冲液,细胞铲刮下细胞(冰上)(3)将裂解液收集到1.5ml离心管中,振荡30s,冰上放置5min,重复2-3次,充分裂解细胞(4)4°C,12000rpm,离心15min,收集上清(5)测定蛋白浓度,取1mg总蛋白做GST-Pull-down?(6)留30μl作为input对照(input与Pull-down蛋白量约为1/10)(7)其余溶液,加入20μl glutathione sepharose beads(PBS洗涤过),4°C,轻柔振荡20min(8)12000rpm,离心3min(9)收集上清,分为两份,一份加入1-15μg GST结合的beads,另一份加入等量GST-融合蛋白结合的beads,4°C,轻柔振荡60min(10)3000rpm,离心3min,回收上清(11)1ml 细胞裂解缓冲液洗涤3次(12)弃尽上清,加入25μl 2×SDS上样缓冲液(13)煮沸3min,12000rpm,离心3min(14)SDS-PAGE,上样顺序:细胞裂解液input、x、GST、x、GST-融合蛋白(x:LSB)(15)考马斯亮蓝染色或免疫印迹[附录]GST裂解缓冲液(前四个组分配好之后4°C保存,DTT和蛋白酶抑制剂现用现加)常用IP细胞裂解液cleared lysates were incubated for 1.5 h with 3 μg immobilized GST or GST-32。