抗肿瘤药达沙替尼的合成进展
新一代抗癌药物的设计与合成

新一代抗癌药物的设计与合成近年来,抗癌药物的设计与合成一直是医药领域的热点研究方向。
随着科技的不断进步和认识的深入,设计出更有效、更安全的新一代抗癌药物成为了许多科学家和研究人员的目标。
本文将介绍新一代抗癌药物的设计与合成的主要方法和技术,为读者提供了解和了解这一领域的基本知识。
一、基于分子靶向的设计方法1. 目标蛋白的筛选与验证在设计新一代抗癌药物时,首先需要确定一个或多个目标蛋白。
通过对癌症相关蛋白的研究和验证,可以选择合适的靶点来设计药物。
这一过程通常包括生物信息学分析、体外实验和转基因动物模型。
2. 靶向结构的筛选与优化一旦确定了目标蛋白,下一步是选择合适的靶向结构。
靶向结构是药物与靶蛋白结合的关键部分,需要具有良好的亲和性和选择性。
通过理论计算和实验方法,可以筛选和优化靶向结构,以提高药物的靶向作用。
3. 药物分子的设计与合成在靶向结构确定后,需要设计并合成药物分子。
这包括选择合适的配体结构和合成路线。
配体结构的设计可以通过分子对接模拟、高通量筛选等方法进行。
合成路线的设计则需要考虑合成的可行性和效率,通常需要经过多步反应来合成目标分子。
二、结构修饰与活性优化1. 结构优化方法对于设计出的初步药物分子,通常需要进行结构修饰和优化,以提高药物的活性和药效。
结构优化可以通过改变配体结构、引入官能团等方法来实现。
这一过程需要结合计算化学和实验测试,寻找最佳的结构修饰方案。
2. 药效评价与活性验证结构修饰后的化合物需要进行药效评价和活性验证。
这包括体外细胞实验和体内动物实验。
通过测定药物对癌细胞的抑制作用、生物转化和毒性等指标,可以评估药物的活性和安全性。
三、新一代抗癌药物的合成方法1. 化学合成方法化学合成是合成新一代抗癌药物的常用方法。
它包括有机合成、无机合成等多种技术和方法。
这些方法通过有机合成反应、金属催化反应等手段,将化学物质按照设计要求,逐步合成出目标药物。
2. 生物合成方法生物合成是另一种重要的合成方法。
新型CDDO-Me类似物的合成及抗肿瘤活性

新型CDDO-Me类似物的合成及抗肿瘤活性乔祎雪;牟伊;黄张建;艾勇;康峰华;赖宜生;张奕华【期刊名称】《中国药科大学学报》【年(卷),期】2015(46)3【摘要】以齐墩果酸(OA)为起始原料,经9步反应合成了C环含有α,β-不饱和酮结构的2-氰基-3,12-二氧代齐墩果烷-1,9(11)-二烯-28-酸甲酯(CDDO-Me)活性类似物1,再将不同的脂肪羧酸和芳杂环羧酸分别与其C-3位羟基酯化,设计、合成了新型CDDO-Me类似物(2a^2e),并进一步将C-1位溴代,得到化合物3a^3e。
采用MTT法测定了目标化合物对肺癌细胞A549、肝癌细胞Hep G2以及肺上皮细胞BEAS-2B的增殖抑制活性。
结果表明,目标化合物对Hep G2细胞和A549细胞的增殖均显示了不同程度的抑制,其中化合物3b和3c的抑制活性最强[IC50=(6.13±1.16)μmol/L,IC50=(5.49±1.03)μmol/L],优于先导物1,与CDDO-Me相当。
此外,目标化合物对正常细胞BEAS-2B的抑制活性显著小于对上述两种肿瘤细胞的抑制活性,显示了较高的肿瘤细胞选择性,其中3e对Hep G2的选择性最高,其抑制作用是正常细胞的10倍,值得进一步研究。
【总页数】5页(P289-293)【关键词】齐墩果酸;CDDO-Me;合成;抗肿瘤活性【作者】乔祎雪;牟伊;黄张建;艾勇;康峰华;赖宜生;张奕华【作者单位】中国药科大学新药研究中心天然药物活性组分与药效国家重点实验室江苏省代谢性疾病药物重点实验室【正文语种】中文【中图分类】R914.5;R965【相关文献】1.抗肿瘤药物达沙替尼类似物的合成及抗肿瘤活性研究 [J], 罗媛;2.新型单羰基姜黄素类似物的合成及抗肿瘤活性研究 [J], 周代营;田宇光;杜志云;赵肃清;郑希;张焜3.齐墩果酸类似物合成及其体外抗肿瘤活性研究 [J], 周颖;孟艳秋4.靶向VEGFR抑制剂齐墩果酸类似物的合成及抗肿瘤活性研究 [J], 高诗特;丁明雪;邬月娇;梅宇;孟艳秋5.积雪草酸新型类似物的合成及体外抗肿瘤活性研究 [J], 林碧琦;孟艳秋因版权原因,仅展示原文概要,查看原文内容请购买。
伊马替尼、达沙替尼及其衍生物的研究

共设计了30个化合物。
一、化合物的设计
一、化合物的设计
二、化合物的合成与结构验证
一共成功合成了20种化合物
二、化合物的合成与结构验证
验证方法: 核磁共振氢谱技术(H NMR):氢原子具有磁性,如电磁波
照射氢原子核,它能通过共振吸收电磁波能量,发生跃迁。用核磁共 振仪可以记录到有关信号,处在不同环境中的氢原子因产生共振时吸 收电磁波的频率不同,在图谱上出现的位置也不同,各种氢原子的这 种差异被称为化学位移。利用化学位移,峰面积和积分值以及耦合常 数等信息,进而推测其在碳骨架上的位置。
伊马替尼、达沙替尼及其衍生物的研究
—— PTKs抑制剂的简单分类举例 & 仿制药研制流程导览
伊马替尼 Imatinib
· 第一个分子靶向治疗的抗肿瘤药
· 分类:Type 2 小分子抑制剂
主要识别激酶的非活性构象,即DFG-out构象。
· 主要识别靶点:Bcr-Abl蛋白激酶
一种非受体型酪氨酸蛋白激酶,由BCR-ABL融合基因表达, 是慢性粒细胞白血病(CML)的重要标志。其通过磷酸化和活化 一系列下游底物,促使CML成熟粒细胞无限增生。BCR-ABL在正 常细胞中不表达,所以它是治疗CML理想的药物靶标。
质谱分析技术(MS):用电场和磁场将运动的离子(带电荷
的原子、分子或分子碎片,有分子离子、同位素离子、碎片离子、重 排离子、多电荷离子、亚稳离子、负离子和离子-分子相互作用产生 的离子)按它们的质荷比分离后进行检测的方法。测出离子准确质量 即可确定离子的化合物组成。
二、化合物的合成与结构验证
二、化合物的合成与结构验证
二、化合物的合成与结构验证
二、化合物的合成与结构验证
三、化合物的活性测定:
上市新药厄达替尼(Erdafitinib)合成检索总结报告
上市新药厄达替尼(Erdafitinib)合成检索总结报告一、厄达替尼(Erdafitinib)简介厄达替尼(Erdafitinib)是由杨森公司研发,并于2019年4月在美国上市抗肿瘤药物。
厄达替尼(Erdafitinib)用于治疗携带特定成纤维细胞生长因子受体(FGFR)基因突变的局部晚期或转移性尿路上皮癌成人患者,具体指携带易感FGFR3或FGFR2基因突变和化疗期间或之后接受至少1种含铂化疗(包括新辅助或辅助含铂化疗12个月内)的局部晚期或转移性尿路上皮癌患者。
厄达替尼作用机制:FGFR激酶抑制剂,可与FGFRl、FGFR2、FGFR3和FGFR4结合并抑制FGFR磷酸化和信号传导,抑制肿瘤细胞的点突变、扩增和融合。
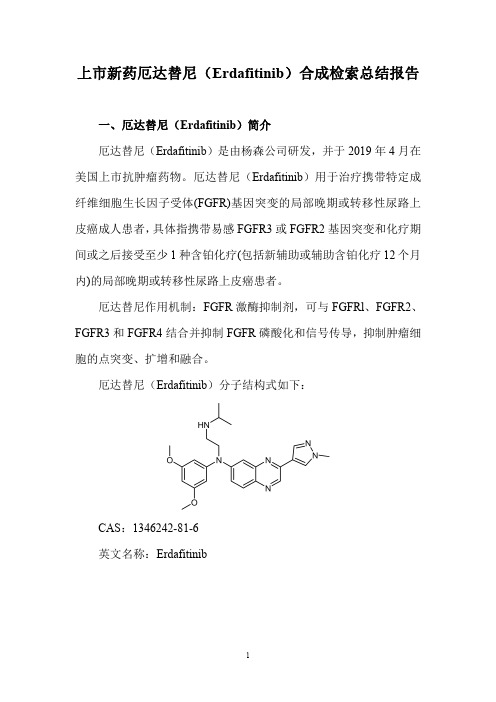
厄达替尼(Erdafitinib)分子结构式如下:CAS:1346242-81-6英文名称:Erdafitinib二、厄达替尼(Erdafitinib)合成路线三、厄达替尼(Erdafitinib )合成检索总结报告(一)厄达替尼中间体2的合成序号实验步骤参考文献1To a suspension of 1,2-diaminobenzene 1(1equiv.)in ethanol (1mol/L)was added ethyl 2-oxoacetate (1.1equiv.).The mixture was stirred at reflux for 1h,then at room temperature overnight.The precipitated solid was filtered and washed with ethanol,then dried to give quinoxalinone 2.Carrer,Amandine;Brion,Jean-Daniel;Messaoudi,Samir;Alami,Mouad;Organic Letters ;vol.15;nb.21;(2013);p.5606-56092To a suspension of o-arylenediamine 1(4.0mmol,1.0equiv)and potassium carbonate (2.0equiv.)in ethanol (1mol/L)was added ethyl 2-oxoacetate (1.1equiv).The reaction mixture was stirred and heated at reflux in an oil bath for 12h,then at room temperature for 12h.Upon completion,the suspension was washed with ethanol,then filtered and dried to give quinoxalinone 2.Noikham,Medena;Kittikool,Tanakorn;Yotphan,Sirilata;Synthesis ;vol.50;nb.12;(2018);p.2337-2346Ethyl 2-oxoacetate (1.1equiv.)was added to a suspension of o -arylenediamine 1(4mmol,1equiv.)in ethanol (1mol/L).The reaction mixture was stirred andSumunnee,Ladawan;Pimpasri,Chaleena;Noikham,Medena;3heated at reflux in anoil bath for 1h,then at room temperature for 16h.Upon completion (as monitored byTLC),the precipitate was filtered and washed with ethanol,then dried to give quinoxalinone 2.Yotphan,Sirilata;Organic and Biomolecular Chemistry ;vol.16;nb.15;(2018);p.2697-27044To a stirred suspension of o-phenylenediamine (50g,462.9mmol)in ethanol(200ml),at rt was added a solution of ethyl glyoxalate in toluene (50;113ml_,555.48mmol)over a period of 45min.After heating to 45°C for 10h,the mixture was left at rt under stirring.The precipitate was filtered and the residue was washed with water and dried to give 1H-quinoxalin-2-one as an off-white powder (63g,93%).WO2011/26579;(2011);(A1)English(二)厄达替尼中间体3的合成序号实验步骤参考文献1To a solution of quinoxalin-2(lH)-one 2(54.64g,374mmol,1.0eq.)in HOAc (1000mL)was added a solution of Br 2(19.18mL,374mmol,1.0eq.)in HOAc (200mL)dropwise.The resulting mixture was stirred at rt for 12h,then poured into ice-water.The precipitate was collected by filtration and dried to afford 7-bromoquinoxalin-2(lH)-one 3as an off-white solid (74g,88%).NEUPHARMA,INC.;QIAN,Xiangping;ZHU,Yong-liang;WO2013/40515;(2013);(A1)2013/53384;(2013);(A1)English 2Quinoxalone 2(250g,1.7mol)is dissolved in acetic acid (4500mL).A mixture of acetic acid (988mL)and bromine (108mL,2.1mol)is added dropwise,and the mixture stirred at room temperature for 12hours,then heated to 60°C for 12hours.After cooling to room temperature,the reaction is filtered and the solid washed with water.The wet cake (500g)is then dissolved in 1500mL of methanol and heated to 60°C,then filtered and dried at 60°C to give 3in 85%yield CLAVIUS PHARMACEUTICALS,LLC;SAWYER,J.,Scott;(109pag.);WO2019/5241;(2019);(A1)English 3To a cooled 0°C solution of quinoxalinone 2(50g,342.2mmol)in acetic acid (800ml)was added in a dropwise manner a solution of bromine (32ml)in acetic acid (200ml_)over a period of 30min.Solids formed within the reaction upon addition of bromine,and the reaction was allowed to stir slowly for a further 90min.WO2016/97918;(2016);(A1)English。
新型靶向抗癌药物的设计与合成研究

新型靶向抗癌药物的设计与合成研究随着科学技术的不断进步和人们对健康的日益关注,抗癌药物的研发变得越来越重要。
近年来,研究人员纷纷将目光投向了新型靶向抗癌药物的设计与合成。
本文旨在探讨新型靶向抗癌药物的设计与合成研究的进展情况,并介绍目前在该领域取得的一些重要成果。
一、新型靶向抗癌药物的定义和意义靶向抗癌药物是指能够选择性地作用于癌细胞,抑制或杀死癌细胞而对正常细胞产生最小损害的药物。
相比于传统的广谱抗癌药物,靶向抗癌药物在治疗效果和副作用上均有明显的优势,因此受到了广泛关注。
靶向抗癌药物的设计与合成是该领域研究的核心内容。
通过精确理解肿瘤发生发展的分子机制,研究人员能够设计出具有高度选择性的药物分子,从而优化抗癌药物的疗效和安全性。
二、新型靶向抗癌药物设计与合成的方法与策略在新型靶向抗癌药物的设计与合成领域,研究人员采用了多种方法和策略。
其中,以下几种是比较常见且有效的方法:1.结构基准法:通过深入了解癌细胞的分子机制,确定潜在的靶点结构。
然后,根据这些结构设计药物分子的结构基准,以实现高度选择性。
2.虚拟筛选法:利用计算机辅助药物设计技术,从大量化合物库中筛选出具有潜在抗肿瘤活性的化合物。
这一方法大大加快了药物研发的速度。
3.合成策略:根据分子机制和目标结构的特点,选择合适的合成路径和合成方法。
有时,还需要进行结构优化和修饰,以提高药物分子的活性和稳定性。
三、新型靶向抗癌药物设计与合成研究的重要成果目前,新型靶向抗癌药物设计与合成研究已经取得了一些重要的成果。
以下是其中的几个代表性成果:1.信号转导抑制剂:这类药物主要通过干扰癌细胞的信号传导通路,抑制癌细胞的生长和增殖。
例如,抑制EGFR (表皮生长因子受体)的分子靶向药物,已经在临床上取得显著的疗效。
2.免疫治疗药物:这类药物主要通过激活或增强人体免疫系统来攻击癌细胞。
近年来,免疫检查点抑制剂的研发取得了突破性进展,部分药物已在多种肿瘤的治疗中得到应用。
达沙替尼作用机制

达沙替尼作用机制达沙替尼是一种靶向治疗药物,主要用于治疗癌症。
它的作用机制是通过抑制特定的信号通路来抑制肿瘤的生长和扩散。
达沙替尼主要作用于细胞中的一种蛋白酪氨酸激酶,称为BCR-ABL 融合蛋白。
BCR-ABL融合蛋白是一种异常的激酶,它的活性过高会导致白血病等恶性肿瘤的发生。
达沙替尼通过与BCR-ABL融合蛋白结合,抑制其活性,从而阻断了信号通路的传导,抑制了肿瘤的生长和扩散。
除了抑制BCR-ABL融合蛋白外,达沙替尼还可以抑制其他一些重要的信号通路。
例如,它可以抑制肿瘤细胞中的血管内皮生长因子受体(VEGFR)和表皮生长因子受体(EGFR),这些受体在肿瘤血管生成和肿瘤细胞增殖中起着重要作用。
通过抑制这些受体,达沙替尼可以减少肿瘤的血供,抑制肿瘤的生长和扩散。
达沙替尼还可以抑制肿瘤细胞中的一种叫做PDGFR的受体。
PDGFR在肿瘤细胞的增殖、生存和迁移中起着重要作用。
通过抑制PDGFR,达沙替尼可以进一步抑制肿瘤的生长和扩散。
总的来说,达沙替尼的作用机制主要包括抑制BCR-ABL融合蛋白、抑制VEGFR、抑制EGFR和抑制PDGFR等。
通过多重靶点的作用,达沙替尼可以同时干扰多个信号通路,从而抑制肿瘤的生长和扩散。
值得注意的是,达沙替尼虽然对肿瘤细胞有较强的抑制作用,但由于其靶向性较强,因此对正常细胞的影响相对较小。
这也是达沙替尼相对传统化疗药物而言的优势之一。
然而,由于每个患者的基因组和疾病特征不同,对达沙替尼的治疗反应也会有差异。
因此,在使用达沙替尼进行治疗时,需要根据患者的具体情况进行个体化的调整和监测。
达沙替尼是一种靶向治疗药物,通过抑制特定信号通路来抑制肿瘤的生长和扩散。
它的作用机制包括抑制BCR-ABL融合蛋白、抑制VEGFR、抑制EGFR和抑制PDGFR等。
通过多重靶点的作用,达沙替尼可以干扰肿瘤细胞的生长和扩散,从而达到治疗癌症的效果。
当然,由于每个患者的情况不同,对达沙替尼的治疗反应也会有所不同,因此需要进行个体化的调整和监测。
新型抗癌药物的合成与活性研究

新型抗癌药物的合成与活性研究近年来,癌症已经成为全球公共卫生问题中备受关注的疾病之一。
随着人类对癌症发生机制的深入研究,越来越多的抗癌药物被发现并运用于临床实践。
但是,现有的抗癌药物还远远不能满足对癌症治疗的需求,这就需要开发新型抗癌药物。
新型抗癌药物的研发离不开化学合成的技术手段。
在新型抗癌药物的合成过程中,要充分考虑其活性和毒性特点。
一方面,药物的活性是癌症治疗的关键,其活性的提高可以增加治疗的效果;另一方面,毒性则需要降低,否则会对患者的身体产生负面影响,而且可能会导致治疗失败。
在新型抗癌药物的研发过程中,需要进行大量的化学合成实验和药效实验。
化学合成实验可以通过改变药物结构,来提高药物的活性和降低药物的毒性。
药效实验则可以评估药物在体内的抗癌效果,为药物的临床应用提供依据。
除了传统的化学合成方法,现在还涌现出了很多新型的合成技术。
其中,基于纳米材料的合成方法日渐发展。
这种方法可以利用纳米材料的特殊性质,在药物的合成和传递方面起到重要作用。
例如,利用金纳米粒子的带电特性,可以把药物冠以金纳米粒子,从而提高药物的生物利用度和稳定性。
另外,生物合成技术也是一种新兴的合成方式。
这种方法利用生物体内的代谢酶合成目标化合物,具有高效、选择性和环境友好等特点。
生物合成技术形成的化合物也可以作为新型抗癌药物的候选物进行突破性研究。
值得注意的是,新型抗癌药物的研发工作和化学、生物、医学等多个领域的前沿研究息息相关。
例如,分子生物学、肿瘤生物学、以及计算机模拟等领域的发展,都为新型抗癌药物的研发提供了有力支持。
总之,新型抗癌药物的合成和活性研究是癌症治疗领域的重要研究方向。
通过不断的创新和实验探索,相信我们一定能够开发出更加有效的抗癌药物,为人类健康事业做出贡献。
CAR-T在恶性肿瘤治疗中的研究进展

CAR-T在恶性肿瘤治疗中的研究进展3空军军医大学药学系生物制药学教研室,西安,710032摘要:CAR-T细胞疗法是一种革命性的癌症治疗方法,它利用改造的T细胞来攻击癌细胞,近年来,CAR-T细胞疗法已经在恶性肿瘤的治疗中取得了显著的进展。
本文通过阐述CAR-T细胞疗法的原理、应用、限制和发展,总结了CAR-T疗法在恶性肿瘤研究中的进展,为未来的研究和临床应用提供了参考。
关键词:CAR-T细胞;恶性肿瘤;淋巴瘤1CAR-T疗法的原理即优势CAR-T(Chimeric Antigen Receptor T-cell)疗法是一种革命性的免疫疗法,用于治疗多种白血病和淋巴瘤等恶性肿瘤,其原理是通过改造患者自身T细胞,使其能够主动攻击癌细胞。
这一疗法的核心是CAR(Chimeric Antigen Receptor)受体,它是一种由合成的受体蛋白构成的分子,能够将T细胞与特定的抗原相结合,从而使T细胞能够识别并杀死癌细胞。
CAR-T疗法的原理可以分为如下几个步骤。
首先是T细胞的采集,医生会从患者的血液中采集T细胞样本,之后,实验室中的研究员会设计和合成CAR受体,这个受体通常包括一个外部抗原结合域、一个跨膜域和一个内部信号传导域。
CAR受体的外部抗原结合域会被设计成能够识别目标癌细胞表面的特定抗原。
然后,采集的T细胞会进行改造,通过病毒载体将CAR受体导入T细胞内部,使其表达CAR受体。
改造后的T细胞会在实验室进行扩增,一旦获得足够数量的CAR-T细胞,它们就会被重新注射入患者的体内,这些细胞就能够识别并攻击患者体内的癌细胞,从而抑制肿瘤的生长和扩散。
CAR-T疗法的优势在于其高度个性化和针对性。
因为CAR-T疗法使用的是患者自身的T细胞,因此每个治疗方案都是高度个性化的,这就意味着CAR-T细胞能够精确地识别和攻击患者体内的癌细胞,最大程度地减少了对健康组织的损害。
此外,CAR-T细胞还具有持久的疗效,一旦它们识别并攻击了癌细胞,就可以在多年后继续活跃并保持疗效。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
l l 4
东
化
工
2 0 1 5年 第 1 5期 第4 2 卷 总第 3 0 5 期
、 ) l n ) . g d c h e m. c o m
臧 佳 良 等 以 2 一 氯。 6 . 甲基 苯胺 为起 始原 料 ,在吡 啶催 化下 与 3 . 乙氧基 丙烯 酰氯 缩 合得 到酰胺 ,再与 N. 溴代 丁二 酰亚 胺和 水 、 硫脲 “ 一 锅法 ” 环 合后 ,在 叔丁 醇 钠 的四氢 呋喃 溶 液 中与 4 , 6 一 二 氯. 2 一 甲基嘧 啶反 应 , 最 后在 三正 丁胺 催化 下 , 与 N. 羟 乙基哌 嗪
一
4 0 f 5 ) :3 2 1 - 3 2 3.
[ 5 ] B a n g - C h i C h e n, R o b e r t o D r o g h i n i ,e t a 1 .P r o c e s s f o r p r e p a r i n g
2 一 m i a n o t h i a z o h . 5 一 a r o ma t i c c a r b o x a mi d e s a s k i n a s e i n h i b i t o r s [ P 1 . U S:
p 5 6 L c k [ J 1 .B i o o r g Me d C h e m L e t t ,2 0 0 6 ,1 3 ( 2 2 ) :4 0 0 7 . 4 0 1 0 . r 3 1 L o mb a r d o L , L e e F Y , C h e n P , e t a 1. D i s c o v e y o r f
/ - -
从 而得 到达 沙替 尼 。 该法 直 接酰 化制 备达 沙 替尼 ,缩 短 了反应 过程 , 并能提 高 收 率 ,而且 2 . 氨基 噻 唑. 3 . 甲酸 乙酯 为起 始 原料 ,价 格低 廉 ,极大 节 约 了成 本 ,工艺 条件 温和 ,操 作简 便 ,适合 工业 生产 。 方法 四:
N一 ( 2 - c h l o r o 一 6 - me f h y l p h e n y 1 ) 一 2 - ( 6 一 ( 4 — 2 - h y d r o x y e t h y 1 ) p i p e r a z i n - 1 ・ y 1 ) 一 2 一 me t h y l p y r i mi d i n - 4 一 y l a mi n o ) t h i a z o l e 一 5 - c a r b o x a mi d e ( B MS - 3 5 4 8 2 5 ) ,a d u a 1 S r c / Ab l k i n a s e i n h i b i t o r wi t h p o t e n t a n t i t u mo r a c t i v i t y i n p r e c l i n i c a l a s s a y s [ J 1 .J Me d C h e m,2 0 0 4 ,4 7 ( 2 7 ) :6 6 5 8 — 6 6 6 1 . [ 4 】 臧 佳 良, 陈一 芬 , 冀亚 飞 . 达沙 替尼 的合 成 [ J ] . 中国医 药工 业杂 志 , 2 0 0 9 ,
N3 o 。.
c・ 。.
。
目
H e l
N
CH3
C h e n B C [ 7 ] 等以 4 . 氨 基一 6 一 氯. 2 . 甲基 嘧啶 为原料 ,先与 异硫 氰 酰 甲酸 乙酯 缩合 , 水解 后 与( E ) 一 N . ( 2 . 氯一 6 一 甲基 苯基) 一 3 . 乙氧 基丙 烯 酰胺 环 合 ,最后 与 N . 羟 乙基 哌嗪 亲核 取代 ,从 而 得到达 沙 替尼 。 该 方法 中用 到 了 4 . 氨基 一 6 . 氯. 2 一 甲基嘧 啶 , 合 成较 困难 , 价 格 昂贵 ,且反 应 的收率 也较 低 ,不利 于工 业生 产 。
方 法三 :
在正 丁醇 中反 应制 得达 沙替 尼 。 该法 的优 点 是反应 工 艺路 线条件 温 和 ,试剂 价廉 易得 ,操 作 简便 ,成 本 不高 ,适合 放大 工业 化生 产 。
( B o c ) 2 0
} f B o c 。 盘
。 .
等
0 ~
张少 宁 等 以 2 一 氨基 噻 唑一 5 . 甲酸 乙酯 为原 料经 过二 碳酸 二叔 丁酯 对氨 基进 行保 护后 ,水 解得 到 2 一 ( N 一 叔 丁氧 羰基 氨 基) . 5 一 噻 唑 甲酸 ,再与草 酰 氯酰 氯化 反应 后无 需减 压蒸 馏 纯化 ,直 接用 于下 步 反应 ,在 二氯 甲烷 的 2 氯一 6 一 甲基苯 胺 的溶 液 中发生 亲核 取代 , 脱去 保护 后 ,与 4 , 6 一 二氯 . 2 . 甲基 嘧啶 及 N- 羟 乙基 哌嗪 亲 核取代 ,
2 结 语
以上各 种不 同合 成 路线 , 基 本上都 是利 用酰 胺化 、 B O C保护 、 水 解 、缩合 、环 合 、取代 反应 。综观 上述 反应 ,有 的 反应步 骤 虽 然 较少 ,例 如方 法 一和 方法 四 ,但其 反应 的原 料很 难购 买 ,反 应 的条件 也 比较 苛刻 , 因此很 难做 到 工业化 生产 。方 法 三 的反应 原 料 易得 ,反应条 件 也 比较温 和 ,但 反应 的步骤 较 长 ,势必会 影 响 产 物 的收率 。综 合 考虑 实际 可操 作性 、成 本 、环 境保 护等 因素 , 笔 者认 为方 法- 为合 成达 沙替 尼较 好 的方法 。