1_3_偶极环加成反应在有机合成方面的新进展
1,3-偶极环加成反应修饰卟啉化合物

维普资讯
第 2 0卷 第 5期
20 0 8年 5月
化 学 进
展
V0 . O No. 12 5
PROGRES N CHEM I T SI S RY
M a ,2 0 y 0 8
13偶极环加成 反应 修饰 卟啉化合物 * ,.
汪 磊 冯 亚 青 一 赵 冰 薛金 强 李 玉坤
Wa g L i F n a i n e eg Yx n lg
Z a ig Xu iqa g L u u hoBn e Jn in i k n Y
(col f hmcl n ne n n eho g ,Taj nvr t,T柚j 00 2 hn ) Sho o e i 西 er gadTcnl y i i U i sy i i 3 0 7 ,C ia C aE i o nn ei n
cc ad i rdc pas pr n r ei po dnmcte p P T a oet ht— nizr rna ic yl d io po ut l i o at l n ht y a i hr y( D ) spt i po s si rf i o tn ya m t o n o a na l o e te o i t a i l
d r aie y t e ra t n fp r h rn d p ls w t i oa p ie uc s C6 ec. i ic s e d t e 1, - ioa e v t sb h ci so o yi io e h dp lr h ls s h a 0 t i v e o p i o s ds u s d a h 3 dp l n r c co d i o fe p d d p r h rn w t zm eh n l e i f re y l a d t n o x a e o yi ih a o ti e yi sr e rd. i n p d e
苯炔在有机合成中的应用

苯炔在有机合成中的应用作者:张志鹏来源:《科学导报·学术》2019年第47期苯炔作为有机化合物中活泼的中间体,反应活性高,在低温条件下就可以发生反应。
苯炔是指缺失两个氢原子的去氢苯,它主要包括三种结构,分别为l,2-去氢苯、l,3-去氢苯、l,4-去氢苯(图1),但一般情况下仅指1,2-去氢苯及其杂环类似物。
它参与的反应大致分为三类:周环反应、亲核加成反应和过渡金属催化的反应。
其中周环反应就可以大概分为四种,主要包括D-A反应;[2+2]环加成反应;l,3-偶极环加成反应;1,4-偶极环加成反应;苯炔参与的ene反应。
苯炔的亲核加成反应分为多种,最主要的是:与氮和碳负离子的亲核加成反应。
苯炔的过渡金属催化反应主要是通过钯催化反应制备多环芳基化合物。
1 周环反应1.1 Diels-Alder反应苯炔中碳碳三键的亲电性很强,在周环反应中可以转化成环戊二烯、呋喃、吡咯、四苯基环戊二烯酮等,然后再与其他试剂发生加成反应。
Snieckus等先用邻硅基芳基磺酸酯生成了中间体苯酰胺苯炔,再将苯酰胺苯炔与呋喃反应,最后得到了产率为63%的环加成的产物。
同样,氨基甲酸酯苯炔也可以与呋喃发生Diels-Alder反应,并且生成与其相似的产物,且产率为79 %,比上述苯酰胺苯炔与呋喃反应的产率都高。
Buszek等人发现,二卤素取代的吲哚,会在叔丁基锂的乙醚溶液中失去卤素原子生成苯炔的衍生物,然后衍生物与呋喃进一步发生加成反应,也会生成萘的衍生物。
Guitián小组将苯炔与两分子的呋喃发生反应,苯炔会与呋喃发生两次Diels-Alder反应的串联过程,然后选择性的得到了多芳环衍生物,再将多芳环衍生物经过酸化等简单操作,最终合成了大稠环芳烃。
李健等以CsF为催化剂,将苯炔在60℃条件下与亚甲基吲哚酮发生反应,在这种反应方法下可以生成含有萘结构的吲哚酮,这种方法在以后制备含萘结构吲哚酮中起到了关键的作用。
有机合成反应的新进展

有机合成反应的新进展近年来,有机合成领域一直在不断推陈出新,为化学界带来了一系列新颖的合成方法和新进展。
本文将介绍一些在有机合成反应中取得的新进展,包括催化剂的设计与应用、绿色合成的发展以及金属有机化学的新突破。
一、催化剂的设计与应用催化剂在有机合成反应中起到了至关重要的作用,能够提高反应速率和选择性。
近年来,科学家们通过对催化剂的设计与优化,取得了一些令人瞩目的成果。
1. 杂环催化剂的应用杂环催化剂是一类具有特殊结构的催化剂,在有机合成领域中得到了广泛应用。
例如,噁唑、噻唑等杂环催化剂能够有效地催化苯胺的C-H活化反应,实现对芳香胺的直接官能团转化。
2. 可持续催化剂的发展随着对环境保护的重视,绿色合成在有机化学中得到了广泛应用。
科学家们致力于开发可持续的催化剂,以减少或避免对环境的污染。
例如,金属有机骨架材料(MOMs)是一种可持续发展的催化剂,具有高效催化性能和可循环利用的特点。
二、绿色合成的发展绿色合成是有机化学合成中的一个热门研究领域,倡导使用环境友好的反应条件和可持续的合成方法。
1. 可再生资源的应用可再生资源是绿色合成的重要组成部分,其利用可以减少对石油等有限资源的依赖。
例如,生物质废弃物可以通过催化转化为有机化学建筑块,再进一步合成有机化合物。
2. 溶剂的选择与优化合理选择溶剂对于绿色合成至关重要。
传统的溶剂如苯、二甲基甲酰胺等对环境有一定的危害。
科学家们通过开发新型溶剂,如离子液体等,取得了可喜的成果。
三、金属有机化学的新突破金属有机化学是有机合成研究的重要分支,通过探索金属有机体系的性质和反应机理,科学家们取得了一些新进展。
1. 金属催化的碳碳键构建金属催化的碳碳键构建反应是有机合成中的重要反应之一。
例如,钯催化的脱氧交叉偶联反应可以实现芳香化合物的构建,极大地拓展了有机合成的可能性。
2. 金属催化的不对称合成不对称合成是现代有机合成领域的热门研究方向。
金属催化的不对称合成反应能够高效地构建手性化合物,对于药物合成和生物活性研究具有重要意义。
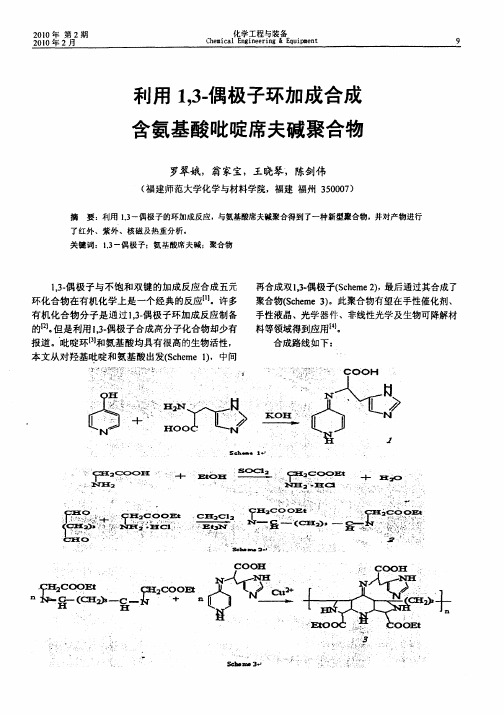
利用1,3-偶极子环加成合成含氨基酸吡啶席夫碱聚合物
。_ ‘
:
‘ :
。
、 。
0
+ 。
u
‘
薯 H ( 、 s )
l H 2 …
・
:
" E6吕 s>ij l t】 t 2 - 【 : 0 ・ c
‘
’
,
. ,
‘ …
●
酸 乙酯盐 酸盐 的制 备 2
铜,7℃反应 6 ,过滤,得到红棕色固体 3 0 h ,产率
6 %。 2
2 结果与讨论
21 F . T—I 分析 ( 1 R 图 ) 图1 是采用 K r 片法 测的产物的红 外谱 图。C B压
在 lO 的三 1瓶 加X 3 0(0 O ml 2 1 . g4 mmo 4 1 氨 酸 )甘
有 机化 合 物 分子 是通 过 l 一 , 偶极 子环 加成 反应制 备 3
再合成 双 1 - 极子(ce e ) 最后通 过其合成 了 ’偶 3 Shm , 2 聚合物(ce ) Shme3 。此聚 合物有望 在手性催 化剂 、
的 。 J但是利用 I . . 偶极子合成高分子化合物却少有 3
02g5 . (mmo) OH 的 4ml 体积无 水 乙醇和蒸馏 8 1 K 0 等
水, 加热至 9 ℃, O 反应数小时, 得黄色溶液, 旋干, 加无水 乙醇后变 混 浊 ,过滤 ,取下层 黄色溶 液,静
置挥发 ,干燥 ,得黄 色固体 1 ,产 率 5%。 3 1 . 1 偶极 子 的合成 .2双 ,3 2
U.. Ni l ga70 SA. c eMan 5傅里 叶变换 红外光谱仪 。 ot
1 . 2合成部分
的戊二醛,然后缓慢滴加含三乙胺( 8 1 1 m) . 的二氯甲 烷溶液2m , 搅拌2 4时,抽滤得红棕色溶液。 0l 4'
1,3-偶极环加成反应-精品文档

BREAD PPT DESIGN
通过2-芳亚甲基-6,7-二氢-5H-噻唑并[3,2-a]嘧啶-3酮与靛红、肌氨酸的l,3-偶极环加成反应,合成了一系列新 的螺噻唑并嘧啶类化合物,以期为药物筛选提供先导化合物。
李筱芳 ,于贤勇 ,冯亚青1,3-偶极环加成反应合成螺噻唑并[3,2-a]嘧啶类化合物, 有机化学2019年第30卷第5期,735-739
BREAD PPT DESIGN
通过六氢-4-芳基-lH-比喃[2,3-d]并嘧啶-2(8aH)硫酮与丁炔二酸二甲(DMAD)的加成反应,合成了一系列新 的吡喃一嘧啶并噻唑类化合物。
姚飞,曾荣今,王慧,沈鹏飞 1,3一偶极环加成反应合成新型的吡喃一嘧啶并噻唑类化 合物 中图分类号:062 文献标识码:A 文章编号:1672—9102(2019)02—0101—04
叠氮基参与 的1,3-偶极 环加成反应 含能盐 含能基团的修饰 新型含能聚合物
池俊杰,夏宇,张晓勤,曲贵晨,常伟林,王建伟 1,3一偶极环加成反应 在含能材料中的应用 中图分类号:0621.3;TJ55 文献标识码:A 文章 编号:1672—2191(2019)03—0025—04
BREAD PPT DESIGN
BREAD PPT DESIGN
叠氮基与碳-碳双 键、碳-碳三键或碳氮三键的1,3一偶极 环加成反应通常有很 多优点。
反应模块化 高产率 反应条件简单 后处理简单
BREAD PPT DESIGN
立体选择性
此类反应是经典点击化学(Click Chemistry) 的精华 。故自2019年诺贝尔化学奖获得者K.B.Sharpless提出 点击化学的概念以来,叠氮基参与的1,3一偶极环加成反 应就在药物合成、分子印迹、超支化聚合物制备、 纳米材料的修饰等众多领域引起了国内外科学家的重视。 含能材料黏合剂
1_3_偶极环加成反应合成1_取代苄基_1_2_3_三唑类化合物

2004年第24卷第10期,1228~1232有机化学Chinese Journal of Organic Chem istryV ol.24,2004N o.10,1228~1232・研究论文・1,32偶极环加成反应合成12(取代苄基)21,2,32三唑类化合物扈艳红Ξ,a 刘世领b 仝钦宇a 黄发荣a沈永嘉b 齐会民a 杜 磊aΞ(a华东理工大学材料科学与工程学院 上海200237)(b华东理工大学精细化工研究所 上海200237)摘要 利用苄氯和取代苄氯与叠氮化钠的亲核取代反应合成了一系列苯环上带有不同取代基团的苄基叠氮化合物,亲核取代反应速率受苯环上取代基的影响:吸电子基团的存在,可以促使反应更容易进行.合成的叠氮化合物与苯乙炔经1,32偶极环加成反应得到了相应的取代苄基1,2,32三唑类化合物,反应条件温和.这些1,2,32三唑类目标化合物具有对热稳定的优点.用红外、核磁、元素分析、质谱等对合成的叠氮化合物和1,2,32三唑类化合物的结构进行了表征,重点研究了1,32环加成反应的规律.加成反应速率取决于叠氮化合物(偶极物)的极性,即与取代基的电负性有关:苯乙炔(亲偶极物)易于与缺电子的叠氮反应,反之亦然.同时在反应过程中观察到空间位阻效应:反应可以生成两种同分异构体,其中42苯基21, 2,32三唑是主要产物.关键词 叠氮,芳基乙炔,1,2,32三唑化合物,1,32偶极环加成Synthesis of12(Substituted benzyl)21,2,32triazoles by1,32Dipolar Cycloaddition R eactionH U,Y an2H ongΞ,a LI U,Shi2Ling b T ONG,Qin2Y u a H UANG,Fa2R ong aSHE N,Y ong2Jia b QI,Hui2Min a DU,Lei a(a School o f Materials Science and Engineering,East China Univer sity o f Science and Technology,Shanghai200237)(b Institute o f Fine Chemicals,East China Univer sity o f Science and Technology,Shanghai200237)Abstract Benzyl azide,42methyl2benzyl azide,42cyano2benzyl azide,42fluoro2benzyl azide and1,1′2bi2 azidomethyl24,4′2biphenyl were prepared by the nucleophilic substitution reaction of NaN3with substituted benzyl chlorides.The reaction rate was affected by the substitution groups at the phenyl ring,and electron2withdrawing groups could accelerate the rate.And1,32dipolar cycloaddition reaction of the azides with phenylacetylene was studied.The corresponding com pounds(azides and1,2,32triazoles)were characterized with NMR,FT2IR and elementary analysis.The1,2,32triazoles were thermal stable.The addition rate depended on the substitution groups of the azides(1,32dipole),and the reactivities correlated with polar effects:phenyacetylene(dipolarophile)reacted faster with electron2poor azides and vice versa.The1,32dipolar cycloaddition reaction was found to be regioselective, and the42phenyl21,2,32triazoles were the major products in the tw o structural is omers of1,2,32triazoles obtained.K eyw ords azide,phenylacetylene,1,2,32triazole,1,32dipolar cycloaddition reaction 叠氮化合物是一类化学性质活泼的化合物,在有机合成和反应机理研究中具有很重要的作用,被广泛地用于合成各种类型的含氮化合物[1~3].20世纪60年代后期,研究发现分子结构中同时具有叠氮基和炔基的双官能团化合物,可以发生分子内的1,32偶极加成反应,生成主链含1,2,32三唑环的线性聚合物,该类型ΞE2mail:fhuanglab@;Fax:021*********.Received October21,2003;revised February23,2003;accepted April19,2004.国家重大基础研究973(N o.5131101)、国家高技术研究发展计划863(N o.2002AA35103)和武器装备预研基金(N o.51412020103QT311)资助项目.的聚合物具有很好的热稳定性.但未见有进一步的后续研究报道[4,5].前苏联利用有机叠氮化合物与多官能团炔烃通过1,32偶极环加成反应,开展合成新型含能粘合剂的研究,国内的北京理工大学等也在开展类似的初步探索[6,7].但是在作为耐高温使用的树脂基复合材料研究领域,国内外目前尚未见有工作报道.在高分子新材料设计与合成中,在聚合物大分子链中引进对称性好的苯环或杂环是获得高耐热聚合物的一条主要途径[8].而1,2,32三唑环由于形成共轭结构,可以形成一类化学性质比较稳定的化合物.基于上述研究设想,我们拟合成一类分子结构中含有1,2,32三唑环的聚合物,希望形成一类新型的耐热聚合物.另一方面,许多涉及叠氮化合物与不饱和烃的1,32偶极加成研究中,作为反应起始物的叠氮化合物主要为脂肪族叠氮衍生物或叠氮基直接与苯环相连的芳香族叠氮衍生物,亲偶极物也主要为脂肪族炔烃[9~12].苯环与叠氮基直接相连的化合物性质不太稳定,有爆炸性,因而限制了它们的实际应用,而相当一部分饱和叠氮化合物和所有含单叠氮基的烷烃的冲击感度都很低[13].因此,本研究拟在苯环与叠氮基团间引入亚甲基,改善芳香叠氮化合物的稳定性,同时,以芳香炔烃替代脂肪族炔烃,探讨环加成反应的可行性和影响因素.通过上述的分子设计,以期在今后聚合物合成过程中,在大分子链中引入对称性好的苯环和三唑杂环,提高聚合物的耐热性能.本工作主要研究了一系列不同取代苄基的叠氮化合物与苯乙炔的1,32偶极环加成反应,探讨了取代基的电负性对反应的影响,得到的目标化合物具有对热稳定的优点,进一步的聚合反应以及应用研究正在进行.叠氮化合物的合成一般采用重氮化反应[11]或卤代烃和叠氮化钠或叠氮化锂反应.然而,上述反应过程较为复杂,特别是重氮化反应条件比较苛刻,产物的提纯以及产率也不高.已有的文献中,大都以溴代烃为原料合成叠氮化物.为在分子结构中引入苯环,同时又降低反应的危险性,本工作以低廉的氯化苄和取代氯化苄为原料,替代通常的溴化物,合成了苄叠氮(3a),对甲基苄叠氮(3b),对氰基苄叠氮(3c),对氟基苄叠氮(3d ),4,4′2联苯二苄叠氮等(3e ′),有效降低反应成本,其中对氰基苄叠氮、对氟基苄叠氮和4,4′2联苯二苄叠氮未见文献报道(Eq.1).叠氮化合物最重要的应用之一是通过1,32偶极环加成反应,与带有不同取代基团的炔烃或烯烃加成反应生成1,2,32三唑或1,2,32三唑啉类的化合物.叠氮基在三个平行的p 轨道中含有4个π电子,称作1,32偶极物,可以与含有2π2电子组分的炔烃或烯烃(称作亲偶极物)发生类似Diels 2Alder 反应的1,32偶极加成反应,生成五元环状化合物1,2,32三唑[14].参照上述原理,利用亲核取代反应制备得到的不同取代基的叠氮化合物与苯乙炔反应,得到加成产物5a ~5d ,6a ~6d 和7(Scheme 1).1 实验红外光谱在NICO LET 5SXC 红外光谱仪上测定;核磁共振氢谱在ADVANCE 500核磁共振仪(美国BRUK A 公司制造)上测得,以T MS 为内标,CDCl 3为溶剂;质谱在HP5989A 质谱仪EI502400(美国HP 公司)上测得;元素分析在德国elementar vario E L III 元素分析仪上测定;熔点在SG W X 24显微熔点仪(上海精密科学仪器有限公司)上测得;热失重分析在DuP ont 1090热失重分析仪(美国DuP ont 公司)上测得,N 2气氛,升温速率10℃/min.Scheme 19221N o.10扈艳红等:1,32偶极环加成反应合成12(取代苄基)21,2,32三唑类化合物 氯化苄,市售分析纯化学试剂;对氰基氯化苄,工业级;对甲基氯化苄,工业级;对氟基氯化苄,工业级;4,4′2联苯二氯化苄,工业级;叠氮化钠,化学纯;苯和DMF均为市售分析纯化学试剂.以上试剂在使用前未作其它处理.苯乙炔采用减压蒸馏进行精制,得到无色透明液体,沸点为142~144℃.1.1 苄基叠氮(3a)的合成在三口烧瓶中加入氯化苄(0105m ol),NaN3(615g,011 m ol),苯(20m L)和DMF(20m L).反应液在搅拌下加热到70℃,在70~75℃保温反应315h.反应进程用薄板层析(T LC)跟踪,反应结束后冷却到室温.将反应液倒入200m L去离子水中,用分液漏斗分出有机相,水相用3×20m L苯萃取,合并有机层,用无水硫酸镁干燥24h,过滤,滤液用旋转蒸发仪除去苯,残余物真空干燥后得淡黄色油状液体,产率68%; 1H NMR(CDCl3)δ:4132(s,2H,CH2),7131~7136(m, 3H,ArH),7137~7141(m,2H,ArH);MS(EI)m/z:133 (M)+,105(M-N2)+,91(M-N3)+,77(M-N3-CH2)+.1.2 对甲基苄叠氮(3b)的合成在三口烧瓶中加入对甲基氯化苄(0105m ol),其余合成方法同111,反应时间415h,产率94%;1H NMR(CDCl3)δ: 2130(s,3H,CH3),4130(s,2H,CH2),7117~7121(m, 4H,ArH);MS(EI)m/z:147(M)+,119(M-N2)+,105 (M-N3)+,91(M-N3-CH2)+.113 对氰基苄叠氮(3c)的合成在三口烧瓶中加入对氰基氯化苄(0105m ol),其余合成方法同111,反应时间215h,产率83%;1H NMR(CDCl3)δ: 4145(s,2H,CH2),7145(d,J=8H z,2H,ArH),7169(t, J=8H z,2H,ArH);MS(EI)m/z:158(M)+,130(M-N2)+,116(M-N3)+,102(M-N3-CH2)+.1.4 对氟基苄叠氮(3d)的合成在三口烧瓶中加入对氟基氯化苄(0105m ol),其余合成方法同1.1,反应时间215h,产率94%;1H NMR(CDCl3)δ: 4130(s,2H,CH2),7104~7109(m,2H,ArH),7124~7129 (m,2H);MS(EI)m/z:151(M)+,123(M-N2)+,109 (M-N3)+,95(M-N3-CH2)+.1.5 4,4′2联苯二苄叠氮(3e′)的合成在三口烧瓶中加入4,4′2联苯二氯化苄(01025m ol),合成方法同1.1,反应时间310h.反应结束将反应液倒入200 m L去离子水中后,静置过夜,析出白色固体.过滤,滤饼用去离子水洗干净,干燥后得白色粉末状固体,产率90%,m.p. 71~72℃;1H NMR(CDCl3)δ:4140(s,2H,CH2),7141(d, J=8H z,2H,ArH),7161(d,J=8H z,2H);MS(EI)m/z: 264(M)+,236(M-N2)+,222(M-N3)+,208(M-N3-CH2)+,180(M-N3-N2-CH2)+,166(M-2N3-CH2)+. 1.6 12苄基242苯基21,2,32三唑(5a)和12苄基252苯基1,2,32三唑(6a)的合成将0101m ol苄基叠氮和0101m ol精制过的苯乙炔加入三口烧瓶中,加入50m L乙醇溶剂回流搅拌下加热28h,反应进程用T LC跟踪(展开剂甲苯∶乙酸乙酯=9∶1,体积比),至反应结束后冷却到室温,用旋转蒸发仪除去溶剂,残余物冷藏析出白色结晶,经过滤后再用乙醚重结晶,得0149g无色针晶,产率21%,柱色谱分离同分异构体(硅胶固定相,洗脱剂:甲苯∶乙酸乙酯=9∶1,体积比),m.p.132~134℃;1H NMR(CDCl3)δ:7183~7175(m,2H,ArH),7166(s,1H, CH),7145~7135(m,5H,ArH),7134~7129(m,3H, ArH),5158(s,2H,CH2);IR(K Br)ν:3120,1467,1223, 1138,973cm-1.Anal.calcd for C13H13N3:C76160,H5153,N 17187;found C76138,H5152,N17189.1.7 12(4′2甲基苄基)242苯基21,2,32三唑(5b)和12(4′2甲基苄基)252苯基21,2,32三唑(6b)的合成在三口瓶中加入0101m ol对甲基苄叠氮,其余合成方法同115,反应时间40h,得0155g无色晶体,产率22%,m.p. 109~111℃;1H NMR(CDCl3)δ:7179(d,J=7134H z,2H, ArH),7163(s,1H,CH),7179(t,J=7158H z,2H, ArH),7131(t,J=7135H z,1H),7121(q,J=8134H z,4H, ArH),5153(s,2H,CH2),2135(s,3H,CH3);IR(K Br)ν: 3115,1463,1223,1133,974cm-1.Anal.calcd for C14H15N3: C77111,H6102,N16187;found C76178,H6105,N16188.1.8 12(4′2氰基苄基)242苯基21,2,32三唑(5c)和12(4′2氰基苄基)252苯基21,2,32三唑(6c)的合成在三口瓶中加入0101m ol对氰基苄叠氮,其余合成方法同115,反应时间23h,得0160g无色晶体,产率23%,m.p. 131~132℃;1H NMR(CDCl3)δ:7187~7177(m,2H, ArH),7170(d,J=8129H z,2H,ArH),7173(s,1H, CH),7148~7132(m,5H,ArH),5170(s,2H,CH2);IR (K Br)ν:3116,1463,1220,1130,974cm-1.Anal.calcd for C16H12N4:C73185,H4162,N21153;found C73142,H4159, N21186.1.9 12(4′2氟苄基)242苯基21,2,32三唑(5d)和12(4′2氟苄基)252苯基21,2,32三唑(6d)的合成在三口瓶中加入0101m ol对氟基苄叠氮,其余合成方法同115,反应时间25h,得0157g无色晶体,产率24%,m.p. 133~135℃;1H NMR(CDCl3)δ:7182~7176(m,2H),7165 (s,1H,CH),7141(t,J=7155H z,2H,ArH),7135~7127(m,3H,ArH),7112~7105(m,2H,ArH),5153(s, 2H,CH2);IR(K Br)ν:3121,1463,1227,1137,974cm-1 (因氟对所测试的元素分析仪有腐蚀作用,故无该化合物元素分析数据).1.10 4,4′2联苯二苄基-二苯基-二21,2,32三唑(7)的合成在三口瓶中加入0105m ol4,4′2联苯二苄叠氮,加入50 m L氯仿回流搅拌,其余合成方法同115,反应时间35h,有淡黄色固体析出.反应结束后,过滤,以氯仿为溶剂,索氏提取器萃取,干燥后得0184g淡黄色固体粉末,产率36%,m.p. 171~173℃;IR(K Br)ν:3124,1461,1219,1184,975 cm-1.Anal.calcd for C30H24N6:C76192,H5113,N17195;0321 有机化学V ol.24,2004found C76179,H5112,N17188.2 结果和讨论2.1 叠氮化合物的合成苄基卤代物与叠氮化钠反应生成苄基叠氮化物的反应是亲核取代反应.因此苯环上的吸电子基团的存在,可以促使反应更容易进行,而供电子基团的存在,则降低反应的速度.氰基和氟原子都是强的吸电子基,因此可以加快反应速度,从实验部分可以看到,氯化苄作为原料的反应时间是315h,以对氰基氯化苄和对氟基氯化苄为原料的反应时间是215h;不仅如此,氰基和氟原子的存在也增加了叠氮化物的稳定性.而供电子基团甲基的存在使对甲基苄叠氮的合成时间为4.5h.2.2 不同取代苄基1,2,32三唑化合物的合成1,32偶极环加成反应类似Diels2Alder反应,与取代苄基叠氮化合物的合成相比较,1,2,32三唑环的生成同样受到1,32偶极物叠氮化合物上取代基团的影响,当带有供电子取代基,如CH3,则反应速度降低,当带有吸电子取代基如CN 或F,则反应加快.实验结果验证了上述设想.T LC跟踪反应进程,对于单官能团化合物可以观察到两个新的产物点出现,柱色谱分离同样得到两种产物.这是因为炔烃的叁键最终加成在1,2,32三唑环的4和5位,炔基上的取代基同样可以在4或5位,生成同分异构体.但是由于空间位阻效应,以苯环在4位的产物为主,比例在60%~90%.不过,每两种同分异构体具有相同的熔点和NMR,FT2 IR图谱,这是由于无论4位或5位上的氢质子,它们的化学环境均非常接近.对1,2,32三唑化合物的红外分析表明,与反应的原料叠氮化合物相比,相应的产物1,2,32三唑化合物中,叠氮基在2100cm-1附近的特征峰完全消失了,也没有苯乙炔2108cm-1的C C拉伸振动的吸收峰和3289cm-1的C—H的拉伸振动,在3100~3200cm-1处出现1,2,32三唑环上C—H 的振动吸收峰,1460cm-1左右出现1,2,32三唑环上氮氮双键的骨架振动特征峰,1220~1227cm-1左右是C—N拉伸振动,1130~1138cm-1归属为N—N摇摆,而974cm-1属于C—H的非平面摇摆振动.红外分析结果初步表明,苯乙炔与叠氮化合物反应,生成1,2,32三唑类化合物.进一步对产物进行元素分析和核磁共振分析,元素分析结果与理论计算值相符.核磁共振分析与产物预计结构可以很好地符合,炔基三键与叠氮基环加成,炔烃上的苯环在1,2,32三唑环上的4或5位.谱图中几乎没有杂峰,表明得到预计产物,而且纯度较高.与文献报道相似结构的化合物数据相比较[15],本实验测得的1,2,32三唑化合物的熔点数据有较大的差距,可能是由于结晶形态不同所致.2.3 模型1,2,32三唑化合物的热分解文献报导分子主链中含1,2,32三唑环的线性聚合物具有较好的热稳定性[4,5],本论文的主要研究目的是合成新型的分子结构中含三唑环的耐热聚合物.利用热失重分析(TG A)考察了模型化合物的热稳定性能.TG A分析数据表明,单官能团的1,2,32三唑化合物中,除CN取代的5c和6c 的混合物的初始热分解温度为32311℃,几乎完全分解温度为36215℃外,其余H,CH3和F取代的5a,6a;5b,6b和5d,6d的热分解温度非常接近,初始热分解温度在24215℃附近,几乎完全分解温度在29715℃附近,我们认为这是因为CN与相连的苄基一起与1,2,32三唑环形成共轭大π键的缘故.而双官能团的化合物7表现出较高的热稳定性,其初始热分解温度为38219℃,几乎完全分解温度为62910℃.我们合成的低聚物的初始热分解温度在350℃以上,而几乎完全分解温度超过了800℃[16].上述结果初步表明,随着分子中1,2,32三唑环数目的增加,化合物表现出良好的耐热性.R eferences1Eric,F.V.S.;K enneth,T.Chem.Rev.1988,88,298.2Zhu,S.2Z.;Xu,Y.;Wang,.Chem.2001,21,986(in Chinese).(朱世正,许勇,王彦利,有机化学,2001,21,986.)3Lw owski,L.In1,32Dipolar Cycloaddition Chemistry,Ed.: Padwa,A.,Wiley,New Y ork,1984,p.559.4Johns on,K. E.;Lovinger,J.A.;Parker,C.O.;Baldwin,M.G.J.Polym.Sci.,Part B:Polym.Lett.1966,4(12),977. 5Baldwin,M.G.;Johns on,K. E.;Lovinger,J.A.;Parker,C.O.J.Polym.Sci.,Part B:Polym.Lett.1967,5(11),803. 6Wang,X.2H.;Feng,Z.2G.;Ling,J.Acta Polym.Sin.2000,(4),397(in Chinese).(王晓红,冯增国,凌剑,高分子学报,2000,(4),397.)7Wang,X.2H.;Feng,Z.2G.Polym.Mater.Sci.Eng.2001, 17(3),1(in Chinese).(王晓红,冯增国,高分子材料科学与工程,2001,17(3),1.)8Lin,S.2A.;Lu,Y.;Liang,Z.2X.In Polymer Chemistry, Science Press,Beijing,2000,p.768(in Chinese).(林尚安,陆耘,梁兆熙,高分子化学,科学出版社,北京, 2000,p.768.)9Francisco,P.;Ana,M.O.de R.;Jaione,P.Heterocycles 1994,38(1),95.10Z anirato,P.J.Chem.Soc.,Perkin Trans.11991,2789.11Chen,M.2D.;Y uan,G.2P.;Y ang,.Chem.2000,20(3),357(in Chinese).(陈敏东,袁光谱,杨世琰,有机化学,2000,20(3),357.) 12Thomas,G.B.;Richard,J. B.;Mas ood,P.;Jerry,A.T.;Daniel,.Chem.1999,64,7426.13Zhou,M.2C.J.Solid Rocket Technol.1990,13(1),59(in Chinese).(周明川,固体火箭技术,1990,13(1),59.)1321N o.10扈艳红等:1,32偶极环加成反应合成12(取代苄基)21,2,32三唑类化合物 14Lw owski ,L.In 1,32Dipolar Cycloaddition Chemistry ,Ed.:Padwa ,A.Wiley ,New Y ork ,1984,p. 3.15Abu 2Orabi ,S.T.;Adnan ,A.M.;Jibril ,I.;Mari ′I ,F.M.;Ali ,A.A.J.Heterocycl.Chem.1989,26,1461.16Hu ,Y.2H.;T ong ,Q.2Y.;Liu ,S.2L.;Huang ,F.2R.;Qi ,H.2M.;Du ,L.In Structures ,Properties and Characterizations o f Polymer s ,2003Chinese Polymer Conference ,Zhejiang University ,Hangzhou ,2003,p.B45(in Chinese ).(扈艳红,仝钦宇,刘世领,黄发荣,齐会民,杜磊,2003全国高分子年会,杭州,2003,p.B45.)(Y 0310218 LI ,L.T.;LI NG,J.)2321 有机化学V ol.24,2004。
通过1,3-偶极环加成反应合成3-吡咯螺环氧化吲哚的研究进展

通过1,3-偶极环加成反应合成3-吡咯螺环氧化吲哚的研究进展周英;张文会;张敏;彭礼军;黄俊飞;杨超;刘雄利;余章彪【摘要】作为一种重要的天然生物碱,吡咯螺环氧化吲哚骨架一直是天然产物化学和药物化学领域里的研究热点.由于含吡咯螺环氧化吲哚骨架分子广泛具有抗氧化、抗肿瘤等生物活性,近年来对其进行全合成和衍生化合成研究也持续升温.以各种取代的氨基酸为原料和各种取代羰基原位产生亚胺叶立德,然后再和α,β-不饱和烯烃发生1,3-偶极[3+2]环加成反应是合成各种吡咯螺环氧化吲哚类化合物的一种有效方法.本文对这一合成方法在近几年的研究进展进行了综述.【期刊名称】《山地农业生物学报》【年(卷),期】2015(034)002【总页数】6页(P9-13,46)【关键词】亚胺叶立德;1,3-偶极环加成反应;吡咯螺环氧化吲哚;综述【作者】周英;张文会;张敏;彭礼军;黄俊飞;杨超;刘雄利;余章彪【作者单位】贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025;贵州大学药学院贵州省中药民族药创制工程中心,贵州贵阳550025【正文语种】中文【中图分类】R914.51 引言由于吡咯螺环氧化吲哚类化合物具有明确或潜在的生物和药物活性,近年来已被大家广泛关注[1]。
例如,spirotryprostatin A(1)[2],pteropodine(2)[3],alstonisine(3)[4],elacomine(4)[5],horsfiline(5)[6],formosanine(6)[7],rychnophylline(7)[8]等都是经典的含具有吡咯螺环氧化吲哚骨架的天然生物碱(见图1)。
1,3偶极环加成合成芳基螺环化合物的研究

( 1新 疆 大学 ,新 疆 乌鲁 木 齐 8 3 0 0 0 2 ; 2杭 州师 范大 学 ,浙江 杭州 3 1 0 0 3 6 )
摘 要 :螺环化合物是一类天然产物和药物中常见的杂环化合物,它们具有多种生物活性,在医学上其抗菌、抗病毒、消
炎 等 生 物 活 性 非 常 显 著 ,是 国 内外 研 究 的一 个 热 点 。1 , 3偶 极 环 加 成 反 应 是 一 个 重 要 的 有 机 合 成 反 应 ,它 的应 用 很 广 泛 ,在 多 种 药物 中间体 的合成 中起 了至关重要 的作用 。本文 以 1 , 3偶极环加成和点击 化学 的方法进 行 了一 系列的芳基螺 环化合物 的合成研究
H E H o n g— q i a n g ,C H A I K e — i f e , XU W e i — ui r n g
( 1 C o l l e g e o f c h e mi s t r y a n d c h e m i c a l i n d u s t r y , X i n j i a n g U n i v e r s i t y , X i n j i a n g U r u m q i 8 3 0 0 0 2 ; 2 C o l l e g e o f Ma t e r i a l C h e mi s t r y a n d C h e mi c a l E n g i n e e r i n g , H a n g z h o u N o r m a l U n i v e r s i t y , Z h e j i a n g H a n g z h o u 3 1 0 0 3 6 ,C h i n a )
第4 2卷第 l 4期