L-Ornithine_70-26-8_DataSheet_MedChemExpress
Genotoxicity A Standard Battery for Genotoxicity Testing

The European Agency for the Evaluation of Medicinal ProductsHuman Medicines Evaluation UnitICH - Technical Coordination - R. Bass 7 Westferry Circus, Canary Wharf, London E14 4HB, UK ICH Topic S 2 BGenotoxicity: A Standard Battery forGenotoxicity Testing of PharmaceuticalsStep 4, Consensus guideline, 16 July 1997NOTE FOR GUIDANCE ON GENOTOXICITY:A STANDARD BATTERY FOR GENOTOXICITY TESTINGOF PHARMACEUTICALS(CPMP/ICH/174/95)TRANSMISSION TO CPMPOctober 1996TRANSMISSION TO INTERESTED PARTIESOctober 1996COMMENTS REQUESTED BEFOREApril 1997FINAL APPROVAL BY CPMPSeptember 1997DATE FOR COMING INTO OPERATION March 1998GENOTOXICITY: A STANDARD BATTERY FORGENOTOXICITY TESTING OF PHARMACEUTICALS[ICH Harmonised Tripartite Guideline]TABLE OF CONTENTS1.INTRODUCTION (2)2.GENERAL PURPOSE OF GENOTOXICITY TESTING (2)3.THE STANDARD TEST BATTERY FOR GENOTOXICITY (2)4.MODIFICATIONS OF THE 3-TEST BATTERY (4)4.1Limitations to the use of bacterial test organisms (4)4.2Compounds bearing structural alerts for genotoxic activity (4)4.3Limitations to the use of standard in vivo tests (4)4.4Additional genotoxicity testing in relation to the carcinogenicity bioassay (5)4.4.1 Evidence for tumour response (5)4.4.2 Structurally unique chemical classes (5)5.STANDARD PROCEDURES FOR IN VITRO TESTS (5)6.NOTES (6)7.GLOSSARY (8)1.INTRODUCTIONTwo fundamental areas in which harmonisation of genotoxicity testing for pharmaceuticals is considered necessary are the scope of this guideline: i) Identification of a standard set of tests to be conducted for registration. ii) The extent of confirmatory experimentation in in vitro genotoxicity tests in the standard battery. Further issues that were considered necessary for harmonisation can be found in the ICH guideline "Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals" (ICH topic S2A). The two ICH guidelines on genotoxicity complement each other and therefore should be used together as ICH guidance principles for testing of a pharmaceutical for potential genotoxicity.2.GENERAL PURPOSE OF GENOTOXICITY TESTINGGenotoxicity tests can be defined as in vitro and in vivo tests designed to detect compounds which induce genetic damage directly or indirectly by various mechanisms. These tests should enable a hazard identification with respect to damage to DNA and its fixation. Fixation of damage to DNA in the form of gene mutations, larger scale chromosomal damage, recombination and numerical chromosome changes is generally considered to be essential for heritable effects and in the multi-step process of malignancy, a complex process in which genetic changes may play only a part. Compounds which are positive in tests that detect such kinds of damage have the potential to be human carcinogens and/or mutagens, i.e. may induce cancer and/or heritable defects. Because the relationship between exposure to particular chemicals and carcinogenesis is established for man, whilst a similar relationship has been difficult to prove for heritable diseases, genotoxicity tests have been used mainly for the prediction of carcinogenicity. Nevertheless, because germ line mutations are clearly associated with human disease, the suspicion that a compound may induce heritable effects is considered to be just as serious as the suspicion that a compound may induce cancer. In addition, the outcome of such tests may be valuable for the interpretation of carcinogenicity studies.3.THE STANDARD TEST BATTERY FOR GENOTOXICITYRegistration of pharmaceuticals requires a comprehensive assessment of their genotoxic potential. It is clear that no single test is capable of detecting all relevant genotoxic agents. Therefore, the usual approach should be to carry out a battery of in vitro and in vivo tests for genotoxicity. Such tests are complementary rather than representing different levels of hierarchy.The general features of a standard test battery can be outlined as follows:i)It is appropriate to assess genotoxicity in a bacterial reverse mutation test. This test hasbeen shown to detect relevant genetic changes and the majority of genotoxic rodent carcinogens.ii)DNA damage considered to be relevant for mammalian cells and not adequately measured in bacteria should be evaluated in mammalian cells. Several mammalian cell systems are in use: systems that detect gross chromosomal damage (in vitro tests forstructural and numerical chromosomal aberrations), systems that detect primarily gene mutations (see Note 1), and a system that detects gene mutations and clastogenic effects (mouse lymphoma tk assay) (see Note 2). The information given in Notes 3 and4 demonstrate that with appropriate test protocols (see Section 5) the various in vitrotests for chromosomal damage and the mouse lymphoma tk assay yield results with a high level of congruence for compounds that are regarded as genotoxic but yield negative results in the bacterial reverse mutation assay. Therefore, these systems are currently considered interchangeable when used together with other genotoxicity tests in a standard battery for genotoxicity testing of pharmaceuticals, if these test protocols are used.iii)An in vivo test for genetic damage should usually be a part of the test battery to provide a test model in which additional relevant factors (absorption, distribution metabolism, excretion) that may influence the genotoxic activity of a compound are included. As a result, in vivo tests permit the detection of some additional genotoxic agents (see Note 5). An in vivo test for chromosomal damage in rodent hematopoietic cells fulfills this need. This in vivo test for chromosomal damage in rodents could be either an analysis of chromosomal aberrations in bone marrow cells or an analysis of micronuclei in bone marrow or peripheral blood erythrocytes.The following standard test battery is recommended based upon the considerations mentioned above:i) A test for gene mutation in bacteria.ii)An in vitro test with cytogenetic evaluation of chromosomal damage with mammalian cells or an in vitro mouse lymphoma tk assay.iii)An in vivo test for chromosomal damage using rodent hematopoietic cells.For compounds giving negative results, the completion of this 3-test battery, performed and evaluated in accordance with current recommendations, will usually provide a sufficient level of safety to demonstrate the absence of genotoxic activity (see Note 6). Compounds giving positive results in the standard test battery may, depending on their therapeutic use, need to be tested more extensively (see ICH Guideline “Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals”).The suggested standard set of tests does not imply that other genotoxicity tests are generally considered as inadequate or inappropriate (e.g. tests for measurement of DNA adducts, DNA strand breaks, DNA repair or recombination). Such tests serve as options in addition to the standard battery for further investigation of genotoxicity test results obtained in the standard battery. Furthermore, molecular techniques to study mechanisms of genotoxicity in the standard battery systems may be useful for risk assessment. Only under extreme conditions in which one or more tests comprising the standard battery cannot be employed for technical reasons, alternative validated tests can serve as substitutes. For this to occur, sufficient scientific justification should be provided to support the argument that a given standard battery test is not appropriate.The standard battery does not include an independent test designed specifically to test for aneuploidy. However, information on this type of damage may be derived from the tests for chromosomal damage in vitro and in vivo. Elements of the standard protocols that provide such information are elevations in the mitotic index, polyploidy induction and micronucleus evaluation. There is also limited experimental evidence that aneuploidy inducers can be detected in the mouse lymphoma tk assay (see Note 4). In such cases, further testing may be needed.4.MODIFICATIONS OF THE 3-TEST BATTERYThe following sections give situations where the standard 3-test battery may need modification.4.1Limitations to the use of bacterial test organismsThere are circumstances where the performance of the bacterial reverse mutation test does not provide appropriate or sufficient information for the assessment of genotoxicity. This may be the case for compounds that are excessively toxic to bacteria (e.g. some antibiotics) and compounds thought or known to interfere with the mammalian cell replication system (e.g. topoisomerase inhibitors, nucleoside analogues, or inhibitors of DNA metabolism). For these cases, usually two in vitro mammalian cell tests should be performed using two different cell types and of two different endpoints [gene mutation (see Note 1) and chromosomal damage]. Nevertheless, it is still important to perform the bacterial reverse mutation test (see Note 7); either a full test or a limited (range-finding) test (see Section 5) may be appropriate.4.2Compounds bearing structural alerts for genotoxic activityStructurally alerting compounds (see Note 8) are usually detectable in the standard 3-test battery. However, compounds bearing structural alerts that have given negative results in the standard 3-test battery may necessitate limited additional testing. The choice of additional test(s) or protocol modification(s) depend on the chemical nature, the known reactivity and metabolism data on the structurally alerting compound under question (see Note 9 and ICH “Guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals”).4.3Limitations to the use of standard in vivo testsThere are compounds for which standard in vivo tests do not provide additional useful information. This includes compounds for which data from studies on toxicokinetics or pharmacokinetics indicate that they are not systemically absorbed and therefore are not available for the target tissues in standard in vivo genotoxicity tests. Examples of such compounds are some radioimaging agents, aluminum based antacids, and some dermally applied pharmaceuticals. In cases where a modification of the route of administration does not provide sufficient target tissue exposure, it may be appropriate to base the evaluation only on in vitro testing.4.4Additional genotoxicity testing in relation to the carcinogenicity bioassay4.4.1Evidence for tumour responseAdditional genotoxicity testing in appropriate models may be conducted for compounds that were negative in the standard 3-test battery but which have shown effects in carcinogenicity bioassay(s) with no clear evidence for a non-genotoxic mechanism. To help understand the mechanism of action, additional testing can include modified conditions for metabolic activation in in vitro tests or can include in vivo tests measuring genetic damage in target organs of tumour induction (e.g. liver UDS test, 32P-postlabelling, mutation induction in transgenes, molecular characterisation of genetic changes in tumour-related genes).4.4.2Structurally unique chemical classesOn rare occasions, a completely novel compound in a unique structural chemical class will be introduced as a pharmaceutical. When such a compound will not be tested in chronic rodent carcinogenicity bioassays, further genotoxicity evaluation may be invoked.5.STANDARD PROCEDURES FOR IN VITRO TESTSReproducibility of experimental results is an essential component of research involving novel methods or unexpected findings; however, the routine testing of chemicals with standard, widely used genotoxicity tests need not always be completely replicated. These tests are sufficiently well characterised and have sufficient internal controls that repetition can usually be avoided if protocols with built-in confirmatory elements, such as those outlined below, are used.For both bacterial and mammalian cell gene mutation tests, the results of a range-finding test can be used to guide the selection of concentrations to be used in the definitive mutagenicity test. By these means, a range-finding test may supply sufficient data to provide reassurance that the reported result is the correct one. In bacterial mutagenicity tests, preliminary range-finding tests performed on all bacterial strains, with and without metabolic activation, with appropriate positive and negative controls, and with quantification of mutants, may be considered a sufficient replication of a subsequent complete test. Similarly, a range-finding test may also be a satisfactory substitute for a complete repeat of a test in gene mutation tests with mammalian cells other than the mouse lymphoma tk assay (see below) if the range-finding test is performed with and without metabolic activation, with appropriate positive and negative controls, and with quantification of mutants (see Note 10).For the cytogenetic evaluation of chromosomal damage in vitro, the test protocol includes the conduct of tests with and without metabolic activation, with appropriate positive and negative controls, where the exposure to the test articles is 3 to 6 hours and a sampling time of approximately 1.5 normal cell cycles from the beginning of the treatment. A continuous treatment without metabolic activation up to the sampling time of approximately 1.5 normal cell cycles is needed in case of a negative result for the short treatment period without metabolic activation. Certain chemicals may be more readily detected by longer treatment or delayed sampling times, e.g. some nucleoside analogues or some nitrosamines. Negative results in the presence of a metabolic activation system may need confirmation on a case by case basis (see Note 11). In any case information on the ploidy status should be obtained byrecording the incidence of polyploid cells as a percentage of the number of metaphase cells. An elevated mitotic index or an increased incidence of polyploid cells may give an indication of the potential of a compound to induce aneuploidy. In such cases, further testing may be needed.For the mouse lymphoma tk assay, the test protocol includes the conduct of tests with and without metabolic activation, with appropriate positive and negative controls, where the exposure to the test articles is 3 to 4 hours. A continuous treatment without metabolic activation for approximately 24 hours is needed in case of a negative result for the short treatment without metabolic activation (see Note 4). Negative results in the presence of a metabolic activation system may need confirmation on a case by case basis (see Note 11). In any case, an acceptable mouse lymphoma tk assay includes (i) the incorporation of positive controls which induces mainly small colonies, (ii) colony sizing for positive controls, solvent controls and at least one positive test compound dose (should any exist), including the culture that gave the greatest mutant frequency.Following such testing, further confirmatory testing in the case of clearly negative or positive test results is not usually needed.Ideally it should be possible to declare test results as clearly negative or clearly positive. However, test results sometimes do not fit the predetermined criteria for a positive or negative call and therefore are declared “equivocal”. The application of statistical methods aids in data interpretation, however, adequate biological interpretation is of critical importance. Nonetheless, further testing is usually indicated for equivocal results.6.NOTES1)Test approaches currently accepted for the assessment of mammalian cell genemutation involve the tk locus using mouse lymphoma L5178Y cells or human lymphoblastoid TK6 cells, the hprt locus using CHO cells, V79 cells, or L5178Y cells, or the gpt locus using AS52 cells.2)The molecular dissection of mutants induced at the tk locus shows a broad range ofgenetic events including point mutations, deletions, translocations, recombinations etc.Small colony mutants have been shown to predominantly lack the tk b allele as a consequence of structural or numerical alterations or recombinational events. There is some evidence that other loci, such as hprt or gpt are also sensitive to large deletion events. However, due to the X-chromosomal origin of the hprt gene which is probably flanked by essential genes, large scale deletionevents or numerical alterations often do not give rise to mutant colonies, thus limiting the sensitivity of this genetic locus relative to the tk locus for the detection of a wide range of genetic changes.3)With respect to the cytogenetic evaluation of chromosomal damage, it is notuncommon for the systems currently in use, i.e. several systems with permanent mammalian cells in culture and human lymphocytes either isolated or in whole blood, to give different results for the same test compound. However, there is evidence that some of the differences observed have been due to protocol differences. This may be minimised by using the procedures described in Section 5.For the great majority of presumptive genotoxic compounds that were negative in a bacterial reverse mutation assay, the data on chromosomal damage in vitro and mouse lymphoma tk results are in agreement. Several reliable studies indicate that the mouse lymphoma tk assay is able to detect compounds that induce structural and numerical chromosomal damage. For safety testing of pharmaceuticals, the mouse lymphoma tk assay is considered an acceptable alternative to the direct analysis of chromosomal damage in vitro. Although colony sizing is an essential element of the mouse lymphoma tk assay test protocol, it gives only limited information on the type of damage induced in mutant colonies. Further mechanistic investigations may be used to assess the nature of cytogenetic changes induced by clastogens and aneuploidy inducers in the mouse lymphoma tk assay. Such information could be provided by studies to demonstrate the loss of the tk gene or the loss of the chromosome carrying the tk gene.4)The detection of a number of different nucleoside analogues and base analogues isenhanced for the mouse lymphoma tk assay when the treatment protocol for both agar and microtitre methods include a 24 hour treatment regimen in the absence of an exogenous metabolic activation system. Similarly, the detection of aneuploidy inducers is enhanced if a 24 hour treatment regimen is used with the microtitre method. Currently, there is no evidence to support this conclusion for the soft agar method. The specificity of the test protocol, i.e. to obtain correct test results for presumptive non-genotoxic compounds, does not change significantly using a 24 hour treatment in the microtitre method. For the soft agar method there appears to be a reduction in specificity under the same treatment regimen. Based on this information, the microtitre method is recommended for use in the standard battery.5)There are a small but significant number of genotoxic carcinogens that are reliablydetected by the bone marrow tests for chromosomal damage that have yielded negative/weak/conflicting results in the pairs of in vitro tests outlined in the standard battery options e.g. bacterial reverse mutation plus one of a selection of possible tests with cytogenetic evaluation of chromosomal damage or bacterial mutation plus the mouse lymphoma tk assay. Carcinogens such as procarbazine, hydroquinone, urethane and benzene fall into this category.6)The continuing evolution of short-term tests and test methodologies will afford new,more sensitive, more practical, more expeditious, and more economical techniques for detection of genotoxic compounds. Some of these may ultimately replace the genotoxicity tests used for regulatory purposes. Among the more promising tests, the in vitro micronucleus test appears to offer potential for screening purposes.7)Some antibacterial agents, albeit highly toxic to the tester strains, are detected asgenotoxic at very low, sub-lethal concentrations in the bacterial reverse mutation test(e.g. nitrofuran antibiotics).8)Certain structurally alerting molecular entities are recognised as being causally relatedto the carcinogenic and/or mutagenic potential of chemicals. Examples of structural alerts include alkylating electrophilic centers, unstable epoxides, aromatic amines, azo-structures, N-nitroso-groups, aromatic nitro-groups.9)For some classes of compounds with specific structural alerts, it is established thatspecific protocol modifications/additional tests are necessary for optimum detection of genotoxicity (e.g. molecules containing an azo-group, glycosides, compounds such as nitroimidazoles requiring nitroreduction for activation, compounds such as phenacetin requiring another rodent S9 for metabolic activation). The additional testing needed when the chosen 3-test battery yields negative results for a structurally alerting test compound could consist of such modifications.10)The dose range-finding study should (i) give information on the shape of the toxicitydose-response curve if the test compound exhibits toxicity, (ii) include highly toxic concentrations, (iii) include quantification of mutants in the cytotoxic range. If a compound were not toxic, then mutants should nevertheless be quantified.11) A repetition of a test using the identical source and concentration of the metabolicactivation system is usually not necessary. A modification of the metabolic activation system may be indicated for certain chemical classes where knowledge is available on specific requirements of metabolism. This would usually invoke the use of an external metabolising system which is known to be competent for the metabolism/activation of the class of compound under test.7.GLOSSARYCytogenetic evaluation: chromosome structure analysis in mitosis or meiosis by light microscopyDNA adduct: (covalent) binding of chemicals to DNADNA repair: reconstitution of damaged DNA sequenceDNA strand breaks: single or double strand scissions in the DNANumerical chromosome changes: chromosome numbers different from the original haploid or diploid set of chromosomes; for cell lines, chromosome numbers different from the modal chromosome setRecombination: breakage and balanced or unbalanced rejoining of DNATransgene: an exogenous or foreign gene inserted into the host genome, either into somatic cells or germ lline cells。
ACCUSPIN系统-Histopaque 1077产品说明书

Technical BulletinACCUSPIN™ System – Histopaque ® 1077Catalog Numbers A6929, A7054, and A0561Product DescriptionACCUSPIN System-Histopaque -1077products are intended for use in the isolation of lymphocytes and other mononuclear cells. The separation medium, Histopaque-1077, is a sterile-filtered, endotoxin tested solution of polysucrose and sodium diatrizoate, adjusted to a density of 1.077 g/mL. The ACCUSPIN tube is specially designed with two chambers separated by a porous high density polyethylene barrier (frit).Separation of lymphocytes and other mononuclear cells from whole blood and bone marrow using density gradientseparation media is based on a published method.1 Histopaque-1077 is suitable for human lymphocyte antigen (HLA) typing 2 and as the initial isolation step prior toenumeration of T, B, and ‘null’ lymphocytes.3 It may also be employed in the preparation of pure lymphocyte suspensions for cell culture and cytotoxicity assays.4ACCUSPIN System-Histopaque-1077 products consist of radiation sterilized polypropylene tubes fitted with a highdensity polyethylene frit and aseptically filled with Histopaque-1077.Histopaque-1077 is a sterile-filtered solution of polysucrose, 57 g/L, and sodium diatrizoate, 90 g/L.Density: 1.076–1.078 g/mL Endotoxin: 0.3 EU/mL pH: 8.8–9.0ACCUSPIN System-Histopaque-1077Catalog No. A692940 × 3 mLEach tube contains 3 mL ofHistopaque 1077-1 and will separate 3-6 mL of anticoagulated blood Catalog No. A7054 12 × 15 mLCatalog No. A0561100 × 15 mLEach tube contains 15 mL ofHistopaque 1077-1 and will separate 15-30 mL of anticoagulated bloodReagents and Equipment Required but Not ProvidedCentrifuge (swinging bucket rotor)capable of generating 100 to 1,000 g Centrifuge tubes for washing mononuclear cellsIsotonic phosphate buffered saline solution or appropriate cell culture mediumPrecautions and DisclaimerFor R&D use only. Not for drug, household, or other uses. Please consult the Safety Data Sheet for information regarding hazards and safe handling practices.Preparation InstructionsSpecimen Collection - Collect blood in preservative-free anticoagulant (EDTA or heparin) or use defibrinated blood. For best results, blood should be processed within 2 hours.On occasion, it may be necessary to dilute the blood sample 3 to 5-fold, depending on absolute cell numbers. A similar volume of prediluted blood may be used or the blood sample may be diluted directly in upper chamber of the ACCUSPIN tube (seeProcedure, step 3). This is appropriate for specimens with hematocrits above normal.Storage/StabilityStore the products at 2–8 C.Histopaque-1077 has an expiration period of 3 years. Reagent label bears expiration date.ProcedureAnticoagulated blood can be added to the top chamber of the tube without risk of mixing with the Histopaque-1077 in the lowerchamber under the frit. On centrifugation the whole blood migrates through the frit to contact with the Histopaque-1077. The elements of greater density displace a volume of Histopaque-1077 above the frit giving a clear separation of the bloodcomponents. The erythrocytes aggregate and the granulocytes become slightly hypertonic, increasing their sedimentation rate, resulting in pelleting at the bottom of the ACCUSPIN Tube. Lymphocytes and other mononuclear cells, e.g., monocytes, remain at the plasma/Histopaque-1077 interface. This dense band of mononuclear cells may be collected by pouring off the contents of the upper chamber or by means of a pipette. Erythrocyte contamination is avoided due to the barrier between the chambers.Most extraneous platelets are removed by low speed centrifugation during the washing steps.1. Bring desired number of tubes to roomtemperature. If Histopaque-1077 isabove the frit prior to use, centrifuge at 1,000 g for 30 seconds at room temperature.Note: Failure to bring ACCUSPIN System-Histopaque-1077 to room temperature may cause limited recovery of mononuclear cells. 2. Label tube(s).3. Freely pour the blood sample into theupper chamber of each ACCUSPIN System-Histopaque-1077 tube.a. Use 3–6 mL of whole blood withACCUSPIN System-Histopaque-1077 tubes, Catalog No. A6929. b. Use 15–30 mL of whole blood withACCUSPIN System-Histopaque-1077 tubes, Catalog Nos. A7054 or A0561. Note: Use of volumes of prediluted or whole blood other than those recommended may result in decreased recovery.4. Centrifuge at 1,000 g for 10 minutes atroom temperature or centrifuge at 800 g for 15 minutes at roomtemperature. Centrifugation at lower temperatures, such as 4 C, may result in cell clumping and poor recovery.Note: If platelet contamination is a concern, add the mononuclear cells to a 4-20% sucrose gradient that has been layered over Histopaque-1077.Centrifuge at 1,000 × g for 10 minutes at room temperature. The platelets will pellet at the bottom, while themononuclear cells will migrate to the Histopaque-1077 layer.5. After centrifugation, carefully aspiratethe plasma layer with a Pasteur pipette to within 0.5 cm of the opaque interface containing mononuclear cells. Properly dispose of the plasma layer.Note: Failure to remove the excesssupernatant may result in contamination of the mononuclear band with plasma proteins.6. Carefully transfer the opaque interfacewith a Pasteur pipette into a clean conical centrifuge tube.Note: Removal of Histopaque-1077 with the mononuclear band increasesgranulocyte contamination from residual granulocytes, which may remain at the mononuclear interface.7. Wash the cells by adding 10 mL ofisotonic phosphate buffered saline solution or appropriate cell culture medium and mix by gently drawing in and out of a Pasteur pipette. 8. Centrifuge at 250 g for 10 minutes. 9. Aspirate the supernatant and discard. 10. Resuspend cell pellet with 5 mL ofisotonic phosphate buffered saline solution or appropriate cell culture medium and mix by gently drawing in and out of a Pasteur pipette.11. Centrifuge at 250 g for 10 minutes. 12. Repeat steps 9, 10, and 11, discardsupernatant and resuspend cell pellet in 0.5 mL of isotonic phosphate buffered saline solution or appropriate cell culture medium. Erythrocytes and granulocytes should pellet to the bottom of the ACCUSPIN tube. Mononuclear cells should band at the interface between the Histopaque-1077 and the plasma. If observed results vary from expected results, please contact Sigma-Aldrich Technical Service for assistance.References1. Boyum, A., Separation of leukocytesfrom blood and bone marrow. Scand. J. Clin. Lab. Invest ., 21 (Suppl 97), 77 (1968).2. Amos, D.B., and Pool, P., “HLA typing” inManual of Clinical Immunology, Rose, N.R., and Friedman, H., eds., American Society for Microbiology, (Washington, DC: 1976) pp. 797-804.3. Winchester, R.J., and Ross, G., “Methodsfor enumerating lymphocyte populations” in Manual of Clinical Immunology, Rose, N.R., and Friedman, H. eds., American Society for Microbiology, (Washington, DC: 1976) pp. 64-76.4. Thorsby, E., and Bratlie, A., “A rapidmethod for preparation of pure lymphocyte suspensions.”Histocompatibility Testing, Terasaki, P.I., ed., 665-666 (1970).The life science business of Merck operates as MilliporeSigma in the U.S. and Canada.Merck, Sigma-Aldrich, ACCUSPIN, and Histopaque are trademarks of Merck KGaA, Darmstadt, Germany or its affiliates. All other trademarks are the property of their respective owners. Detailed information on trademarks is available via publicly accessible resources.© 2022 Merck KGaA, Darmstadt, Germany and/or its affiliates. All Rights Reserved.NoticeWe provide information and advice to our customers on application technologies and regulatory matters to the best of our knowledge and ability, but without obligation or liability. Existing laws and regulations are to be observed in all cases by our customers. This also applies in respect to any rights of third parties. Our information and advice do not relieve our customers of their own responsibility for checking the suitability of our products for the envisaged purpose.The information in this document is subject to change without notice and should not be construed as a commitment by the manufacturing or selling entity, or an affiliate. We assume no responsibility for any errors that may appear in this document.Contact InformationFor the location of the office nearest you, go to /offices .Technical ServiceVisit the tech service page on our web site at /techservice .Standard WarrantyThe applicable warranty for the products listed in this publication may be found at /terms .A0561 Technical Bulletin Rev 06/2022。
超高效液相色谱-串联质谱法测定人血浆中精氨酸及衍生物含量

超高效液相色谱-串联质谱法测定人血浆中精氨酸及衍生物含量田晔;江骥;胡蓓;薛金萍;王洪允【摘要】建立了超高效液相色谱-串联质谱(UPLC-MS/MS)法同时测定使用艾普拉唑后人血浆中二甲基精氨酸(ADMA)、对称二甲基精氨酸(SDMA)、单甲基精氨酸(NMMA)、瓜氨酸(Cit)和L-精氨酸(L-Arg)的浓度.采用HILIC亲水相互作用色谱和非衍生化的蛋白沉淀法进行分离分析,色谱柱选取Waters Atlantic HILIC柱(2.1 mm×50 mm×3μm),流动相由乙腈(含0.5%乙酸和0.025%三氟乙酸)-水(含0.5%乙酸和0.025%三氟乙酸)(85:15,v/V)组成,流速0.25 mL/min.采用多反应离子监测(MRM)模式,以电喷雾离子源(ESI)正离子方式检测.结果显示,ADMA、SDMA、NMMA、L-Arg和Cit的线性关系良好,相关系数r均大于0.994 0;ADMA、SDMA和NMMA的线性范围为0.1~5 mmol/L,L-Arg和Cit的线性范围为10~250 mmol/L;5种氨基酸的日内、日间精密度均小于15%,准确度在85%~115%之间.该方法快速、简便、灵敏,可为相关疾病的临床诊断提供一种高效的检测手段.【期刊名称】《质谱学报》【年(卷),期】2016(037)005【总页数】7页(P446-452)【关键词】超高效液相色谱-串联质谱(UPLC-MS/MS);艾普拉唑;蛋白沉淀法;亲水性色谱【作者】田晔;江骥;胡蓓;薛金萍;王洪允【作者单位】福州大学化学学院,福建省功能材料工程研究中心,福建省光动力治疗药物与诊疗工程技术研究中心,福建福州350108;中国医学科学院北京协和医院临床药理中心,北京100730;中国医学科学院北京协和医院临床药理中心,北京100730;中国医学科学院北京协和医院临床药理中心,北京100730;福州大学化学学院,福建省功能材料工程研究中心,福建省光动力治疗药物与诊疗工程技术研究中心,福建福州350108;中国医学科学院北京协和医院临床药理中心,北京100730【正文语种】中文【中图分类】O657.63一氧化氮是人体重要的信使分子,L-精氨酸(L-Arg)在一氧化氮全酶(NOS)的催化下,产生一氧化氮(NO)和瓜氨酸(Cit)[1-2]。
Antibody structure, instability, and formulation

MINIREVIEWAntibody Structure,Instability,and FormulationWEI WANG,SATISH SINGH,DAVID L.ZENG,KEVIN KING,SANDEEP NEMAPfizer,Inc.,Global Biologics,700Chesterfield Parkway West,Chesterfield,Missouri63017Received14March2006;revised17May2006;accepted4June2006Published online in Wiley InterScience().DOI10.1002/jps.20727 ABSTRACT:The number of therapeutic monoclonal antibody in development hasincreased tremendously over the last several years and this trend continues.At presentthere are more than23approved antibodies on the US market and an estimated200ormore are in development.Although antibodies share certain structural similarities,development of commercially viable antibody pharmaceuticals has not been straightfor-ward because of their unique and somewhat unpredictable solution behavior.This articlereviews the structure and function of antibodies and the mechanisms of physical andchemical instabilities.Various aspects of formulation development have been examinedto identify the critical attributes for the stabilization of antibodies.ß2006Wiley-Liss,Inc.and the American Pharmacists Association J Pharm Sci96:1–26,2007Keywords:biotechnology;stabilization;protein formulation;protein aggregation;freeze drying/lyophilizationINTRODUCTIONProtein therapies are entering a new era with the influx of a significant number of antibody pharmaceuticals.Generally,protein drugs are effective at low concentrations with less side effects relative to small molecule drugs,even though,in rare cases,protein-induced antibody formation could be serious.1Therefore,this category of therapeutics is gaining tremendous momentum and widespread recognition both in small and large drugfirms.Among protein drug therapies,antibodies play a major role in control-ling many types of diseases such as cancer, infectious diseases,allergy,autoimmune dis-eases,and inflammation.Since the approval of thefirst monoclonal antibody(MAb)product -OKT-3in1986,more than23MAb drug products have entered the market(Tab.1).The estimated number of antibodies and antibody derivatives constitute20%of biopharmaceutical products currently in development(about200).2The global therapeutic antibody market was predicted to reach$16.7billion in2008.3There are several reasons for the increasing popularity of antibodies for commercial develop-ment.First,their action is specific,generally leading to fewer side effects.Second,antibodies may be conjugated to another therapeutic entity for efficient delivery of this entity to a target site, thus reducing potential side effects.For instance, Mylotarg is an approved chemotherapy agent composed of calicheamicin conjugated to huma-nized IgG4,which binds specifically to CD33for the treatment of CD33-positive acute myeloid leukemia.Another example is the conjugation of immunotoxic barnase with the light chain of the anti-human ferritin monoclonal antibody F11as potential targeting agents for cancer immuno-therapy.4Third,antibodies may be conjugated to radioisotopes for specific diagnostic purposes. Examples include CEA-Scan for detection of color-ectal cancer and ProstaScint for detection of prostate stly,technology advancement has made complete human MAb available,which are lessimmunogenic.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY20071 Correspondence to:Wei Wang(Telephone:(636)-247-2111;Fax:(636)-247-5030;E-mail:wei.2.wang@pfi)Journal of Pharmaceutical Sciences,Vol.96,1–26(2007)Pharmacists AssociationT a b l e 1.C o m m e r c i a l M o n o c l o n a l A n t i b o d y P r o d u c t s#B r a n d n a m e M o l e c u l eM A bY e a r C o m p a n y R o u t e I n d i c a t i o n M A b C o n c B u f f e r E x c i p i e n t s S u r f a c t a n t p H1A v a s t i n B e v a c i z u m a bH u m a n i z e d I g G 1,149k D a2004G e n e t e c h a n d B i o O n c o l o g y I V i n f u s i o nM e t a s t a t i c c a r c i n o m a o f c o l o n o r r e c t u m ,b i n d s V E G F 100m g a n d 400m g /v i a l (25m g /m L )s o l u t i o n 5.8m g /m L m o n o b a s i c N a P h o s H 2O ;1.2m g /m L d i b a s i c N a P h o s a n h y d r o u s (4m L ,16m L fil l i n v i a l )60m g /m L a -T r e h a l o s e d i h y d r a t e (4m L ,16m L fil l i n v i a l )0.4m g /m L P S 20(4m L ,16m L fil l i n v i a l )6.22B e x x a rT o s i t u m o m a b a n d I -131T o s i t u m a b M u r i n e I g G 2l2003C o r i x a a n d G S KI V I n f u s i o nC D 20p o s i t i v e f o l l i c u l a r n o n H o d g k i n s l y m p h o m aK i t :14m g /m L M A b s o l u t i o n i n 35m g a n d 225m g v i a l s ;1.1m g /m L I 131-M A b s o l u t i o n10m M p h o s p h a t e (M A b v i a l )145m M N a C l ,10%w /v M a l t o s e ;I 131-M A b :5–6%P o v i d o n e ,1–2,9–15m g /m L M a l t o s e ,0.9m g /m L N a C l ,0.9–1.3m g /m L A s c o r b i c a c i d 7.23C a m p a t h A l e m t u z u m a bH u m a n i z e d ,I g G 1k ,150k D a2001I l e x O n c o l o g y ;M i l l e n i u m a n d B e r l e xI V i n f u s i o nB -c e l l c h r o n i c l y m p h o c y t i c l e u k e m i a ,CD 52-a n t i g e n 30m g /3m L s o l u t i o n3.5m g /3m L d i b a s i c N a P h o s ,0.6m g /3m L m o n o b a s i c K P h o s 24m g /3m L N a C l ,0.6m g /3m L K C l ,0.056m g /3m L N a 2E D T A 0.3m g /3m L P S 806.8–7.44C E A -S c a n (l y o )A c r i t u o m a b ;T c -99M u r i n e F a b ,50k D a1996I m m u n o m e d i c s I V i n j e c t i o n o r i n f u s i o nI m a g i n g a g e n t f o r c o l o r e c t a l c a n c e r1.25m g /v i a l L y o p h i l i z e d M A b .R e c o n s t i t u t e w 1m L S a l i n e w T c 99m 0.29m g /v i a l S t a n n o u s c h l o r i d e ,p o t a s s i u m s o d i u m t a r t r a t e t e t r a h y d r a t e ,N a A c e t a t e .3H 2O ,N a C l ,g l a c i a l a c e t i c a c i d ,H C l S u c r o s e5.75E r b i t u x C e t u x i m a bC h i m e r i c h u m a n /m o u s e I g G 1k ,152kD a 2004I m C l o n e a n d B M S I V i n f u s i o n T r e a t m e n t o fE GF R -e x p r e s s i n g c o l o r e c t a l c a r c i n o m a 100m g M A b i n 50m L ;2m g /m L s o l u t i o n1.88m g /m L D i b a s i c N a P h o s Á7H 2O ;0.42m g /m L M o n o b a s i c N a P h o s ÁH 2O8.48m g /m L N a C l 7.0–7.46H e r c e p t i n (l y o )T r a s t u z u m a bH u m a n i z e d I g G 1k1998G e n e t e c h I V i n f u s i o n M e t a s t a t i c b r e a s t c a n c e r w h o s e t u m o r o v e r e x p r e s s H E R 2p r o t e i n 440m g /v i a l ,21m g /m L a f t e r r e c o n s t i t u t i o n 9.9m g /20m L L -H i s t i d i n e H C l ,6.4m g /20m L L -H i s t i d i n e400m g /20m L a -T r e h a l o s e D i h y d r a t e 1.8m g /20m L P S 2067H u m i r a A d a l i m u m a bH u m a n I g G 1k ,148k D a2002C A T a n d A b b o t t S CR A p a t i e n t s n o t r e s p o n d i n g t o D M A R D s .B l o c k s T N F -a l p h a40m g /0.8m L s o l u t i o n (50m g /m L )0.69m g /0.8m L M o n o b a s i c N a P h o s Á2H 2O ;1.22m g /0.8m L D i b a s i c N a P h o s Á2H 2O ;0.24m g /0.8m L N a C i t r a t e ,1.04m g /0.8m L C i t r i c a c i d ÁH 2O 4.93m g /0.8m L N a C l ;9.6m g /0.8m L M a n n n i t o l 0.8m g /0.8m L P S 805.28L u c e n t i s R a n i b i z u m a bH u m a n i z e d I g G 1k f r a g m e n t2006G e n e n t e c h I n t r a v i t r e a l i n j e c t i o n A g e -r e l a t e d m a c u l a r d e g e n e r a t i o n (w e t )10m g /m L s o l u t i o n10m M H i s t i d i n e H C l10%a -T r e h a l o s e -D i h y d r a t e 0.01%P S 205.52WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 2007DOI 10.1002/jps9M y l o t a r g (l y o )G e m t u z u m a b o z o g a m i c i nH u m a n i z e d I g G 4k c o n j u g a t e d w i t h c a l i c h e a m i c i n2000C e l l t e c h a n d W y e t h I V i n f u s i o nH u m a n i z e d A b l i n k e d t o c a l i c h e a m i c i n f o r t r e a t m e n t o f C D 33p o s i t i v e a c u t e m y e l o i d l e u k e m i a 5m g p r o t e i n -e q u i v a l e n t l y o p h i l i z e d p o w d e r /20-m L v i a l M o n o b a s i c a n d d i b a s i c N a P h o s p h a t e D e x t r a n 40,S u c r o s e ,N a C l 10O n c o S c i n tS a t u m o m a b p e n d e t i d eM u r i n e I g G 1k c o n j u g a t e d t o G Y K -D T P A1992C y t o g e n I V i n j e c t i o nI m a g i n g a g e n t f o r c o l o r e c t a l a n d o v a r i a n c a n c e r0.5m g c o n j u g a t e /m L s o l u t i o n (2m L p e r v i a l )P h o s p h a t e b u f f e r s a l i n e 6.011O r t h o c l o n e O K TM u r o m o m a b -C D 3M u r i n e ,I g G 2a ,170k D a1986O r t h o B i o t e c h I V i n j e c t i o nR e v e r s a l o f a c u t e k i d n e y t r a n s p l a n t r e j e c t i o n (a n t i C D 3-a n t i g e n )1m g /m L s o l u t i o n2.25m g /5m L m o n o b a s i c N a P h o s ,9.0m g /5m L d i b a s i c N a P h o s 43m g /5m L N a C l 1m g /m L P S 807Æ0.512P r o s t a S c i n tI n d i u m -111c a p r o m a b p e n d e t i d e M u r i n e I g G 1k -c o n j u g a t e d t o G Y K -D T P A1996C y t o g e n I V i n j e c t i o nI m a g i n g a g e n t f o r p r o s t a t e c a n c e r0.5m g c o n j u g a t e /m L s o l u t i o n (1m L p e r v i a l )P h o s p h a t e b u f f e r s a l i n e 5–713R a p t i v a (l y o )E f a l i z u m a bH u m a n i z e d I g G 1k2003X o m a a n d G e n e n t e c h S C C h r o n i c m o d e r a t e t o s e v e r e p l a q u e p s o r i a s i s ,b i n d s t o C D 11a s u b u n i t o f L F A -1150m g M A b /v i a l ;125m g /1.25m L (100m g /m L )a f t e r r e c o n s t i t u t i o n w i t h 1.3m L S W F I 6.8m g /v i a l L -H i s t i d i n e H C l ÁH 2O ;4.3m g /v i a l L -H i s t i d i n e123.2m g /v i a l S u c r o s e 3m g /v i a l P S 206.214R e m i c a d e (l y o )I n fli x i m a bC h i m e r i c h u m a n /m u r i n e M A b a g a i n s t T N F a l p h a (a p p .30%m u r i n e ,70%c o r r e s p o n d s t o h u m a n I g G 1h e a v y c h a i n a n d h u m a n k a p p a l i g h t c h a i n c o n s t a n t r e g i o n s )1998C e n t o c o r I V i n f u s i o nR A a n d C r o h n ’s d i s e a s e (a n t i T N F a l p h a )100m g /20-m L V i a l ,10m g /m L o n r e c o n s t i t u t i o n2.2m g /10m L M o n o b a s i c N a P h o s H 2O ,6.1m g /10m L D i b a s i c N a P h o s Á2H 2O 500m g /10m L S u c r o s e 0.5m g /10m L P S 807.215R e o P r o A b c i x i m a bF a b .C h i m e r i c h u m a n -m u r i n e ,48k D a 1994C e n t o c o r /L i l l y I V i n j e c t i o n a n d i n f u s i o n R e d u c t i o n o f a c u t e b l o o d c l o t r e l a t e d c o m p l i c a t i o n s 2m g /m L s o l u t i o n 0.01M N a P h o s p h a t e 0.15M N a C l 0.001%(0.01m g /m L )P S 807.216R i t u x a n R i t u x i m a bC h i m e r i c m o u s e /h u m a n I g G 1k w i t h m u r i n e l i g h t a n d h e a v y c h a i n v a r i a b l e r e g i o n (F a b d o m a i n ),145kD a1997I D E C a n d G e n e n t e c h I V i n f u s i o nN o n H o d g k i n ’s l y m p h o m a .(a n t i C D 20-a n t i g e n )10m g /m L s o l u t i o n7.35m g /m L N a C i t r a t e Á2H 2O9m g /m L N a C l 0.7m g /m L P S 806.5(C o n t i n u e d )ANTIBODY FORMULATION3DOI 10.1002/jpsJOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 200717S i m u l e c t (l y o )B a s i l i x i m a bC h i m a r i c I g G 1k ,144kD a1998N o v a r t i s I V i n j e c t i o n a n d i n f u s i o nP r e v e n t i o n o f a c u t e k i d n e y t r a n s p l a n t r e j e c t i o n ,I L -2r e c e p t o r a n t a g o n i s t10m g a n d 20m g /v i a l ,4m g /m L o n r e c o n s t i t u t i o n 3.61m g ,7.21m g M o n o b a s i c K P h o s ;0.50m g ,0.99m g N a 2H P O 40.8m g ,1.61m g N a C l ;10m g ,20m g S u c r o s e ;40m g ,80m g M a n n i t o l ;20m g 40m g G l y c i n e 18S y n a g i s (l y o )P a l i v i z u m a bH u m a n i z e d I g G 1k ,C D R o f m u r i n e M A b 1129,148k D a 1998M e d I m m u n e I M i n j e c t i o nP r e v e n t r e p l i c a t i o n o f t h e R e s p i r a t o r y s y n c y t i a l v i r u s (R S V )50m g a n d 100m g /v i a l ,100m g /m L o n r e c o n s t i t u t i o n47m M H i s t i d i n e ,3.0m M G l y c i n e 5.6%M a n n i t o l19T y s a b r i N a t a l i z u m a bH u m a i n z e d I g G 4k2004B i o g e n I D E C I V I n f u s i o nM S r e l a p s e 300m g /15m L s o l u t i o n 17.0m g M o n o b a s i c N a P h o s ÁH 2O ,7.24m g d i B a s i c N a P h o s Á7H 2O f o r 15m L 123m g /15m L N a C l3.0m g /15m L P S 806.120V e r l u m a N o f e t u m o m a b M u r i n e F a b 1996B o e h r i n g e r I n g e l h e i m a n d D u P o n t M e r c k I V i n j e c t i o n I m a g i n g a g e n t f o r l u n g c a n c e r10m g /m L s o l u t i o nP h o s p h a t e b u f f e r s a l i n e?21X o l a i r (l y o )O m a l i z u m a bH u m a n i z e d I g G 1k ,149k D aG e n e n t e c h w N o v a r t i s a n d T a n o xS CA s t h m a ,i n h i b i t s b i n d i n g o f I g E t o I g E r e c e p t o r F C e R I202.5m g /v i a l ,D e l i v e r 150m g /1.2m L o n r e c o n s t i t u t i o n w i t h 1.4m L S W F I 2.8m g L H i s t i d i n e H C l ÁH 2O ;1.8m g L H i s t i d i n e145.5m g S u c r o s e 0.5m g P S 2022Z e n a p a x D a c l i z u m a bH u m a n i z e d I g G 1,144k D a1997R o c h e I V i n f u s i o nP r o p h y l a x i s o f a c u t e o r g a n r e j e c t i o n i n p a t i e n t s r e c e i v i n g r e n a l t r a n s p l a n t s .I n h i b i t s I L -2b i n d i n g t o t h e T a c s u b u n i t o f I L -2r e c e p t o r c o m p l e x 25m g /5m L M A b S o l u t i o n3.6m g /m L M o n o b a s i c N a P h o s ÁH 2O ;11m g /m L D i b a s i c N a P h o s Á7H 2O4.6m g /m L N a C l 0.2m g /m L P S 806.923Z e v a l i nI b r i t u m o m a b -T i u x e t a nM u r i n e I g G 1k -t h i o u r e a c o v a l e n t l i n k a g e t o T i u x e t a nI D E C I V i n f u s i o nC D 20a n t i g e n .(K i t w i t h Y t t e r i u m -90i n d u c e s c e l l u l a r d a m a g e b y b e t a e m i s s i o n )3.2m g /2m L s o l u t i o n 09%N a C l 7.1T a b l e 1.(C o n t i n u e d )#B r a n d n a m e M o l e c u l eM A bY e a r C o m p a n y R o u t e I n d i c a t i o n M A b C o n c B u f f e r E x c i p i e n t s S u r f a c t a n t p H4WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY 2007DOI 10.1002/jpsDevelopment of commercially viable antibody pharmaceuticals has,however,not been straight-forward.This is because the behavior of antibodies seems to vary,even though they have similar structures.In attempting to address some of the challenges in developing antibody therapeutics, Harris et al.5reviewed the commercial-scale formulation and characterization of therapeutic recombinant antibodies.In a different review, antibody production and purification have been discussed.2Nevertheless,the overall instability and stabilization of antibody drug candidates have not been carefully examined in the litera-ture.This article,not meant to be exhaustive, intends to review the structure and functions of antibodies,discuss their instabilities,and sum-marize the methods for stabilizing/formulating antibodies.ANTIBODY STRUCTUREAntibodies(immunoglobulins)are roughly Y-shaped molecules or combination of such molecules(Fig.1). Their structures are divided into two regions—the variable(V)region(top of the Y)defining antigen-binding properties and the constant(C)region (stem of the Y),interacting with effector cells and molecules.Immunoglobulins can be divided into five different classesÀIgA,IgD,IgE,IgM,and IgG based on their C regions,respectively desig-nated as a,d,e,m,and g(five main heavy-chain classes).6Most IgGs are monomers,but IgA and IgM are respectively,dimmers and pentamers linked by J chains.IgGs are the most abundant,widely used for therapeutic purposes,and their structures will be discussed as antibody examples in detail.Primary StructureThe structure of IgGs have been thoroughly reviewed.6The features of the primary structure of antibodies include heavy and light chains, glycosylation,disulfide bond,and heterogeneity. Heavy and Light ChainsIgGs contain two identical heavy(H,50kDa)and two identical light(L,25kDa)chains(Fig.1). Therefore,the total molecular weight is approxi-mately150kDa.There are several disulfide bonds linking the two heavy chains,linking the heavy and light chains,and residing inside the chains (also see next section).IgGs are further divided into several subclasses—IgG1,IgG2,IgG3,and IgG4(in order of relative abundance in human plasma),with different heavy chains,named g1, g2,g3,and g4,respectively.The structural differences among these subtypes are the number and location of interchain disulfide bonds and the length of the hinge region.The light chains consist of two types—lambda(l)and kappa(k). In mice,the average of k to l ratio is20:1,whereas it is2:1in humans.6The variable(V)regions of both chains cover approximately thefirst 110amino acids,forming the antigen-binding (Fab)regions,whereas the remaining sequences are constant(C)regions,forming Fc(fragment crystallizable)regions for effector recognition and binding.6The N-terminal sequences of both the heavy and light chains vary greatly between different antibodies.It was suggested that the conserved sequences in human IgG1antibodies Figure1.Linear(upper panel)and steric(lower panel)structures of immunoglobulins(IgG).ANTIBODY FORMULATION5DOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007are approximately95%and the remaining5% is variable and creates their antigen-binding specificity.5The V regions are further divided into three hypervariable sequences(HV1,HV2,and HV3)on both H and L chains.In the light chains,these are roughly from residues28to35,from49to59,and from92to103,respectively.6Other regions are the framework regions(FR1,FR2,FR3,and FR4).The HV regions are also called the complementarity determining regions(CDR1,CDR2,and CDR3). While the framework regions form the b-sheets, the HV sequences form three loops at the outer edge of the b barrel(also see Section2.2).Disulfide BondsMost IgGs have four interchain disulfide bonds—two connecting the two H chains at the hinge region and the other two connecting the two L chains to the H chains.6Exceptions do exist.Two disulfide bonds were found in IgG1and IgG4 linking the two heavy chain in the hinge region but four in IgG2.7In IgG1MAb,HC is linked to the LC between thefifth Cys(C217)of HC and C213on the LC.In IgG2and IgG4MAbs,it is the third Cys of HC(C123)linking to the LC.7A disulfide bond between HC C128and LC C214 was found for mouse catalytic monoclonal anti-bodies(IgG2a).8IgGs have four intrachain disulfide bonds, residing in each domain of the H and L chains, stabilizing these domains.The intrachain disul-fide bonds in V H and V L are required in functional antigen binding.9Native IgG MAbs should not have any free sulfhydryl groups.7However, detailed examination of the free sulfhydryl groups in recombinant MAbs(one IgG1,two IgG2,and one IgG4)suggests presence of a small portion of free sulfhydryl group(approximately0.02mol per mole of IgG2or IgG4MAb and0.03for IgG1.7In rare cases,a free cysteine is found.A nondisulfide-bonded Cys at residue105was found on the heavy chain of a mouse monoclonal antibody,OKT3 (IgG2a).10OligosaccharidesThere is one oligosaccharide chain in IgGs.6This N-linked biantennary sugar chain resides mostly on the conserved Asn297,which is buried between the C H2domains.5,11For example,the oligosaccharide resides on Asn-297of the C H2 domain of chimeric IgG1and IgG3molecules12but on Asn299in a monoclonal antibody,OKT3 (IgG2a).10The oligosaccharide,often microheter-ogeneous,is typically fucosylated in antibodies produced in CHO or myeloma cell lines5and may differ in other cell lines.2,11There are many factors that dictate the nature of the glycan microheterogenity on IgGs.These include cell line,the bioreactor conditions and the nature of the downstream processing.An additional oligo-saccharide can be found in rare cases.A human IgG produced by a human-human-mouse hetero-hybridoma contains an additional oligosaccharide on Asn75in the variable region of its heavy chain.13In addition,O-linked carbohydrates could also exist in this antibody.Proper glycosylation is critical for correct functioning of antibodies.11It was demonstrated that removal of the oligosaccharide in IgGs(IgG1 and IgG3)made them ineffective in binding to C1q, in binding to the human Fc g RI and activating C; and generally more sensitive to most proteases than their corresponding wild-type IgGs(one exception).12This is because the binding site on IgG for C1q,thefirst component of the complement cascade,is localized in the C H2domains.11 Furthermore,the glycosylation can affect the antibody conformation.12Oligosaccharides in other regions can also play a critical role.Removal of an oligosaccharide in a Fv region of the CBGA1antibody resulted in a decreased antigen-binding activity in several ELISA systems.13In addition,this oligosaccharide might play critical role in reducing the antigenicity of the protein.14The sugar composition of the oligosaccharide is also critical in antibody functions.It has been shown that a low fucose(Fuc)content in the complex-type oligosaccharide in a humanized chimeric IgG1is responsible for a50-fold higher antibody-dependent cellular cytotoxicity(ADCC) compared with a high Fuc counterpart.15 HeterogeneityPurified antibodies are heterogeneous in struc-ture.This is true for all monoclonal antibodies (MAbs)due to differences in glycosylation pat-terns,instability during production,and terminal processing.5For example,five charged isoforms were found in recombinant humanized monoclo-nal antibody HER2as found by capillary iso-electric focusing(cIEF)and sodium dodecyl sulfate–capillary gel electrophoresis(SDS–CGE).16Six separate bands were focused under6WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007DOI10.1002/jpsIEF for two mouse monoclonal antibodies IgG2a (k)and IgG1(k).17A mature monoclonal antibody, OKT3(IgG2a),contain cyclized N-terminus (pyroglutamic acid,À17D)in both H and L chains, processed C-terminus(no Lys,À128D)of the H chains,and a small amount of deamidated form.10 Similar observation was also reported for a huma-nized IgG1(k).18In rare cases,gene cross-over may lead to formation of abnormal heavy chains.For example,a purified monoclonal anti-IgE antibody contains a small amount of a variant H chain, which had16fewer amino acid residues than the normal H chain(position is between Arg108of the L chain and Ala124of the H chain).19 Secondary and Higher-Order StructureThe basic secondary and higher-order structural features of IgGs have been reviewed.6Only a small portion of the three-dimensional structures of IgGs has been solved.20The antibody’s secon-day structure is formed as the polypeptide chains form anti-parallel b-sheets.The major type of secondary structure in IgGs is these b-sheets and its content is roughly70%as measured by FTIR.21The light chain consists of two and the heavy chain contains four domains,each about 110amino acid long.6,20All these domains have similar folded structures—b barrel,also called immunoglobulin fold,which is stabilized by a disulfide bond and hydrophobic interaction(pri-mary).These individual domains($12kDa in size)interact with one another(V H and V L;C H1 and C L;and between two C H3domains except the carbohydrate-containing C H2domain)and fold into three equal-sized spherical shape linked by a flexible hinge region.These three spheres form a Y shape(mostly)and/or a T shape.22The less globular shape of IgGs is maintained both by disulfide bonds and by strong noncovalent interactions between the two heavy chains and between each of the heavy-chain/light-chain pairs.23Through noncovalent interactions,a less stable domain becomes more stable,and thus,the whole molecule can be stabilized.24A detailed study indicates that the interaction between two CH3domains are dominated by six contact residues,five of these residues(T366,L368, F405,Y407,and K409)forming a patch at the center of the interface.25These noncovalent interactions are spatially oriented such that variable domain exchange(switching V H and V L; inside-out IgG;ioIgG)induces noncovalent multimerization.26The six hypervariable regions in CDR(L1,L2, L3,H1,H2,and H3)form loops of a few predictable main-chain conformations(or canonical forms), except H3loop,which has too many variations in conformation to be predicted accurately.27,28 There is a slight difference in the loop composition and shape between the two types of light chains.20 However,no functional difference was found in antibodies having l or k chain.6Basic Functions of AntibodiesThe basic functions of antibodies have been reviewed.6There are two functional areas in IgGs—the V and C regions.The V regions of the two heavy and light chains offer two identical antigen-binding sites.The binding of the two sites (bivalent)can be independent of each other and does not seem to depend on the C region.29The exact antigen-binding sites are the CDR regions with participation of the frame work regions.30 Binding of antigens seems through the induced-fit mechanism.31,32The induced-fit mechanism allows multispecificity and polyreactivity.It has been suggested that about5–10residues usually contribute significantly to the binding energy.32 The C regions of antibodies have three main effector functions(1)being recognized by receptors on immune effector cells,initiating antibody-dependent cell cytotoxicities(ADCC),(2)binding to complement,helping to recruit activated pha-gocytes,and(3)being transported to a variety of places,such as tears and milk.6In addition,C domains also modulate in vivo stability.23,29,33The function of Fc is affected by the structure of Fab. Variable domain exchange(switching V H and V L; inside-out IgG;ioIgG)affected Fc-associated func-tions such as serum half-life and binding to protein G and Fc g RI.26The hinge region providesflexibility in bivalent antigen binding and activation of Fc effector functions.26Two chimeric IgG3antibodies lacking a genetic hinge but with Cys residues in CH2 regions was found to be deficient in their inter-molecular assembly,and both IgG3D HþCys and IgG3D Hþ2Cys lost greatly their ability to bind Fc g RI and failed to bind C1q and activate the complement cascade.34Alternative Forms of AntibodiesIn addition to species-specific antibodies,other antibody forms are generated to meet various needs.In the early development of antibody therapies,antibodies were made from murineANTIBODY FORMULATION7DOI10.1002/jps JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007sources.However,these antibodies easily elicit formation of human anti-mouse antibody (HAMA).Therefore,humanized chimeric antibo-dies were generated.Chimeric monoclonal anti-bodies(60–70%human)are made of mouse variable regions and human constant regions.2 Such antibodies can still induce formation of human anti-chimeric antibody(HACA).Highly humanized antibodies,CDR-grafted antibodies, are made by replacing only the human CDR with mouse CDR regions(90–95%human).2These antibodies are almost the same in immunogeni-city potential as completely human antibodies, which may illicit formation of human anti-human antibody(HAHA).Other alternative forms of antibodies have also been generated and these different forms have been reviewed.35Treatment with papain would cleave the N-terminal side of the disulfide bonds and generate two identical Fab fragments and one Fc fragment.Fab0s are50kDa(V HþC H1)/ (V LþC L)heterodimers linked by a single disul-fide bond.Treatment with pepsin cleaves the C-terminal side of the disulfide bonds and pro-duces a F(ab)02fragment.The remaining H chains were cut into several small fragments.6Cleavage by papain occurs at the C-terminal side of His-H22836or His-H227.37Reduction of F(ab0)2will produce two Fab0.23Fv fragments are noncovalent heterodimers of V H and V L.Stabilization of the fragment by a hydrophilicflexible peptide linker generates single-chain Fv(scFvs).2Fragments without constant domains can also be made into domain antibodies (dAbs).These scFvs are25–30kDa variable domain (V HþV L)dimers joined by polypeptide linkers of at least12residues.Shorter linkers(5–10residues)do not allow pairing of the variable domains but allow association with another scFv form a bivalent dimer (diabody)(about60kDa,or trimer:triabody about 90kDa).38Two diabodies can be further linked together to generate bispecific tandem diabody (tandab).39Disulfide-free scFv molecules are rela-tively stable and useful for intracellular applica-tions of antibodies—‘‘intrabodies.’’38The smallest of the antibody fragments is the minimal recognition unit(MRU)that can be derived from the peptide sequences of a single CDR.2ANTIBODY INSTABILITYAntibodies,like other proteins,are prone to a variety of physical and chemical degradation path-ways,although antibodies,on the average,seem to be more stable than other proteins.Antibody instabilities can be observed in liquid,frozen,and lyophilized states.The glycosylation state of an antibody can significantly affect its degradation rate.40In many cases,multiple degradation path-ways can occur at the same time and the degrada-tion mechanism may change depending on the stress conditions.41These degradation pathways are divided into two major categories—physical and chemical instabilities.This section will explore the possible degradation pathways of antibodies and their influencing factors.Physical InstabilityAntibodies can show physical instability via two major pathways—denaturation and aggregation. DenaturationAntibodies can denature under a variety of conditions.These conditions include temperature change,shear,and various processing steps. Compared with other proteins,antibodies seem to be more resistant to thermal stress.They may not melt completely until temperature is raised above708C,21,42,43while most other mesophilic proteins seem to melt below708C.44Shear may cause antibody denaturation.For example,the antigen-binding activity of a recombinant scFv antibody fragment was reduced with afirst-order rate constant of0.83/h in a buffer solution at a shear of approximately20,000/s.45Lyophilization can denature a protein to var-ious extents.An anti-idiotypic antibody(MMA 383)in a formulation containing mannitol,sac-charose,NaCl,and phosphate was found to loose its in vivo immunogenic properties(only10–20% of normal response rate)upon lyophilization.46 Since the protein showed no evidence of degrada-tion after lyophilization,no change in secondary structure by CD(29%b-sheet,14%a-helix,and 57%‘‘other’’),the loss of activity was attributed to the conformational change.Indeed,tryptophan fluorescence properties were different between the lyophilized and unlyophilized antibodies.46 AggregationAntibody aggregation is a more common manifes-tation of physical instability.The concentration-dependent antibody aggregation was considered the greatest challenge to developing protein formulations at higher concentrations.47This is8WANG ET AL.JOURNAL OF PHARMACEUTICAL SCIENCES,VOL.96,NO.1,JANUARY2007DOI10.1002/jps。
marked manuscript

Quality evaluation of Flos Lonicerae through a simultaneous determination of seven saponins by HPLC with ELSDXing-Yun Chai1, Song-Lin Li2, Ping Li1*1Key Laboratory of Modern Chinese Medicines and Department of Pharmacognosy, China Pharmaceutical University, Nanjing, 210009, People’s Republic of China2Institute of Nanjing Military Command for Drug Control, Nanjing, 210002, People’s Republic of China*Corresponding author: Ping LiKey Laboratory of Modern Chinese Medicines and Department of Pharmacognosy, China Pharmaceutical University, Nanjing 210009, People’s Republic of China.E-mail address: lipingli@Tel.: +86-25-8324-2299; 8539-1244; 135********Fax: +86-25-8532-2747AbstractA new HPLC coupled with evaporative light scattering detection (ELSD) method has been developed for the simultaneous quantitative determination of seven major saponins, namely macranthoidinB (1), macranthoidin A (2), dipsacoside B (3), hederagenin-28-O-β-D-glucopyranosyl(6→1)-O-β-D- glucopyranosyl ester (4), macranthoside B (5), macranthoside A (6), and hederagenin-3-O-α-L-arabinopyranosyl(2→1)-O-α-L-rhamnopyranoside (7)in Flos Lonicerae, a commonly used traditional Chinese medicine (TCM) herb.Simultaneous separation of these seven saponins was achieved on a C18 analytical column with a mixed mobile phase consisting of acetonitrile(A)-water(B)(29:71 v/v) acidified with 0.5% acetic acid. The elution was operated from keeping 29%A for 10min, then gradually to 54%B from 10 to 25 min on linear gradient, and then keep isocratic elution with 54%B from 25 to 30min.The drift tube temperature of ELSD was set at 106℃, and with the nitrogen flow-rate of 2.6 l/min. All calibration curves showed good linear regression (r2 0.9922) within test ranges. This method showed good reproducibility for the quantification of these seven saponins in Flos Lonicerae with intra- and inter-day variations of less than 3.0% and 6.0% respectively. The validated method was successfully applied to quantify seven saponins in five sources of Flos Lonicerae, which provides a new basis of overall assessment on quality of Flos Lonicerae.Keywords: HPLC-ELSD; Flos Lonicerae; Saponins; Quantification1. IntroductionFlos Lonicerae (Jinyinhua in Chinese), the dried buds of several species of the genus Lonicera (Caprifoliaceae), is a commonly used traditional Chinese medicine (TCM) herb. It has been used for centuries in TCM practice for the treatment of sores, carbuncles, furuncles, swelling and affections caused by exopathogenic wind-heat or epidemic febrile diseases at the early stage [1]. Though four species of Lonicera are documented as the sources of Flos Lonicerae in China Pharmacopeia (2000 edition), i.e. L. japonica, L. hypoglauca,L. daystyla and L. confusa, other species such as L. similes and L. macranthoides have also been used on the same purpose in some local areas in China [2]. So it is an important issue to comprehensively evaluate the different sources of Flos Lonicerae, so as to ensure the clinical efficacy of this Chinese herbal drug.Chemical and pharmacological investigations on Flos Lonicerae resulted in discovering several kinds of bioactive components, i.e. chlorogenic acid and its analogues, flavonoids, iridoid glucosides and triterpenoid saponins [3]. Previously, chlorogenic acid has been used as the chemical marker for the quality evaluation of Flos Lonicerae,owing to its antipyretic and antibiotic property as well as its high content in the herb. But this compound is not a characteristic component of Flos Lonicerae, as it has also been used as the chemical marker for other Chinese herbal drugs such as Flos Chrysanthemi and so on[4-5]. Moreover, chlorogenic acid alone could not be responsible for the overall pharmacological activities of Flos Lonicerae[6].On the other hand, many studies revealed that triterpenoidal saponins of Flos Lonicerae possess protection effects on hepatic injury caused by Acetaminophen, Cd, and CCl4, and conspicuous depressant effects on swelling of ear croton oil [7-11]. Therefore, saponins should also be considered as one of the markers for quality control of Flos Lonicerae. Consequently, determinations of all types of components such as chlorogenic acid, flavonoids, iridoid glucosides and triterpenoidal saponins in Flos Lonicerae could be a better strategy for the comprehensive quality evaluation of Flos Lonicerae.Recently an HPLC-ELSD method has been established in our laboratory for qualitative and quantitative determination of iridoid glucosides in Flos Lonicerae [12]. But no method was reported for the determination of triterpenoidal saponins in Flos Lonicera. As a series studies on the comprehensive evaluation of Flos Lonicera, we report here, for the first time, the development of an HPLC-ELSD method for simultaneous determination of seven triterpenoidal saponins in the Chinese herbal drug Flos Lonicerae, i.e.macranthoidin B (1), macranthoidin A (2), dipsacoside B (3), hederagenin-28-O-β-D-glucopyranosyl(6→1)-O-β-D- glucopyranosyl ester (4), macranthoside B (5), macranthoside A (6), and hederagenin-3-O-α-L-arabinopyranosyl(2→1)-O-α-L-rhamnopyranoside (7) (Fig. 1).2. Experimental2.1. Samples, chemicals and reagentsFive samples of Lonicera species,L. japonica from Mi county, HeNan province (LJ1999-07), L. hypoglauca from Jiujang county, JiangXi province (LH2001-06), L. similes from Fei county, ShanDong province (LS2001-07), L. confuse from Xupu county, HuNan province (LC2001-07), and L. macranthoides from Longhu county, HuNan province (LM2000-06) respectively, were collected in China. All samples were authenticated by Dr. Ping Li, professor of department of Pharmacognosy, China Pharmaceutical University, Nanjing, China. The voucher specimens were deposited in the department of Pharmacognosy, China Pharmaceutical University, Nanjing, China. Seven saponin reference compounds: macranthoidin B (1), macranthoidin A (2), dipsacoside B (3), hederagenin-28-O-β-D-glucopyranosyl(6→1)-O-β-D- glucopyranosyl ester (4), macranthoside B (5), macranthoside A (6), and hederagenin-3-O-α-L-arabinopyranosyl(2→1)-O-α-L-rhamnopyranoside (7) were isolated previously from the dried buds of L. confusa by repeated silica gel, sephadex LH-20 and Rp-18 silica gel column chromatography, their structures were elucidated by comparison of their spectral data (UV, IR, MS, 1H- NMR and 13C-NMR) with references [13-15]. The purity of these saponins were determined to be more than 98% by normalization of the peak areas detected by HPLC with ELSD, and showed very stable in methanol solution.HPLC-grade acetonitrile from Merck (Darmstadt, Germany), the deionized water from Robust (Guangzhou, China), were purchased. The other solvents, purchased from Nanjing Chemical Factory (Nanjing, China) were of analytical grade.2.2. Apparatus and chromatographic conditionsAglient1100 series HPLC apparatus was used. Chromatography was carried out on an Aglient Zorbax SB-C18 column(250 4.6mm, 5.0µm)at a column temperature of 25℃.A Rheodyne 7125i sampling valve (Cotati, USA) equipped with a sample loop of 20µl was used for sample injection. The analog signal from Alltech ELSD 2000 (Alltech, Deerfield, IL, USA)was transmitted to a HP Chemstation for processing through an Agilent 35900E (Agilent Technologies, USA).The optimum resolution was obtained by using a linear gradient elution. The mobile phase was composed of acetonitrile(A) and water(B) which acidified with 0.5% acetic acid. The elution was operated from keeping 29%A for 10min, then gradually to 54%B from 10 to 25 min in linear gradient, and back to the isocratic elution of 54%B from 25 to 30 min.The drift tube temperature for ELSD was set at 106℃and the nitrogen flow-rate was of 2.6 l/min. The chromatographic peaks were identified by comparing their retention time with that of each reference compound tried under the same chromatographic conditions with a series of mobile phases. In addition, spiking samples with the reference compounds further confirmed the identities of the peaks.2.3. Calibration curvesMethanol stock solutions containing seven analytes were prepared and diluted to appropriate concentration for the construction of calibration curves. Six concentrationof the seven analytes’ solution were injected in triplicate, and then the calibration curves were constructed by plotting the peak areas versus the concentration of each analyte. The results were demonstrated in Table1.2.4. Limits of detection and quantificationMethanol stock solution containing seven reference compounds were diluted to a series of appropriate concentrations with methanol, and an aliquot of the diluted solutions were injected into HPLC for analysis.The limits of detection (LOD) and quantification (LOQ) under the present chromatographic conditions were determined at a signal-to-noise ratio (S/N) of 3 and 10, respectively. LOD and LOQ for each compound were shown in Table1.2.5. Precision and accuracyIntra- and inter-day variations were chosen to determine the precision of the developed assay. Approximate 2.0g of the pulverized samples of L. macranthoides were weighted, extracted and analyzed as described in 2.6 Sample preparation section. For intra-day variability test, the samples were analyzed in triplicate for three times within one day, while for inter-day variability test, the samples were examined in triplicate for consecutive three days. Variations were expressed by the relative standard deviations. The results were given in Table 2.Recovery test was used to evaluate the accuracy of this method. Accurate amounts of seven saponins were added to approximate 1.0g of L. macranthoides,and then extracted and analyzed as described in 2.6 Sample preparation section. The average recoveries were counted by the formula: recovery (%) = (amount found –original amount)/ amount spiked ×100%, and RSD (%) = (SD/mean) ×100%. The results were given in Table 3.2.6. Sample preparationSamples of Flos Lonicerae were dried at 50℃until constant weight. Approximate 2.0g of the pulverized samples, accurately weighed, was extracted with 60% ethanol in a flask for 4h. The ethanol was evaporated to dryness with a rotary evaporator. Residue was dissolved in water, followed by defatting with 60ml of petroleum ether for 2 times, and then the water solution was evaporated, residue was dissolved with methanol into a 25ml flask. One ml of the methanol solution was drawn and transferred to a 5ml flask, diluted to the mark with methanol. The resultant solution was at last filtrated through a 0.45µm syringe filter (Type Millex-HA, Millipore, USA) and 20µl of the filtrate was injected to HPLC system. The contents of the analytes were determined from the corresponding calibration curves.3. Results and discussionsThe temperature of drift tube and the gas flow-rate are two most important adjustable parameters for ELSD, they play a prominent role to an analyte response. In ourprevious work [12], the temperature of drift tube was optimized at 90°C for the determination of iridoids. As the polarity of saponins are higher than that of iridoids, more water was used in the mobile phase for the separation of saponins, therefore the temperature for saponins determination was optimized systematically from 95°C to 110°C, the flow-rate from 2.2 to 3.0 l/min. Dipsacoside B was selected as the testing saponin for optimizing ELSD conditions, as it was contained in all samples. Eventually, the drift tube temperature of 106℃and a gas flow of 2.6 l/min were optimized to detect the analytes. And these two exact experimental parameters should be strictly controlled in the analytical procedure [16].All calibration curves showed good linear regression (r2 0.9922) within test ranges. Validation studies of this method proved that this assay has good reproducibility. As shown in Table 2, the overall intra- and inter-day variations are less than 6% for all seven analytes. As demonstrated in Table 3, the developed analytical method has good accuracy with the overall recovery of high than 96% for the analytes concerned. The limit of detection (S/N=3) and the limit of quantification (S/N=10) are less than 0.26μg and 0.88μg respectively (Table1), indicating that this HPLC-ELSD method is precise, accurate and se nsitive enough for the quantitative evaluation of major non- chromaphoric saponins in Flos Lonicerae.It has been reported that there are two major types of saponins in Flos Lonicerae, i.e. saponins with hederagenin as aglycone and saponins with oleanolic acid as the aglycone [17]. But hederagenin type saponins of the herb were reported to have distinct activities of liver protection and anti-inflammatory [7-11]. So we adoptedseven hederagenin type saponins as representative markers to establish a quality control method.The newly established HPLC-ELSD method was applied to analyze seven analytes in five plant sources of Flos Lonicerae, i.e. L. japonica,L. hypoglauca,L. confusa,L. similes and L. macranthoides(Table 4). It was found that there were remarkable differences of seven saponins contents between different plant sources of Flos Lonicerae. All seven saponins analyzed could be detected in L. confusa and L. hypoglauca, while only dipsacoside B was detected in L. japonica. Among all seven saponins interested, only dipsacoside B was found in all five plant species of Flos Lonicerae analyzed, and this compound was determined as the major saponin with content of 53.7 mg/g in L. hypoglauca. On the other hand, macranthoidin B was found to be the major saponin with the content higher than 41.0mg/g in L. macranthoides,L. confusa, and L. similis, while the contents of other analytes were much lower.In our previous study [12], overall HPLC profiles of iridoid glucosides was used to qualitatively and quantitatively distinguish different origins of Flos Lonicerae. As shown in Fig.2, the chromatogram profiles of L. confusa, L. japonica and L. similes seem to be similar, resulting in the difficulty of clarifying the origins of Flos Lonicerae solely by HPLC profiles of saponins, in addition to the clear difference of the HPLC profiles of saponins from L. macranthoides and L. hypoglauca.Therefore, in addition to the conventional morphological and histological identification methods, the contents and the HPLC profiles of saponins and iridoids could also be used as accessory chemical evidence toclarify the botanical origin and comprehensive quality evaluation of Flos Lonicerae.4. ConclusionsThis is the first report on validation of an analytical method for qualification and quantification of saponins in Flos Lonicerae. This newly established HPLC-ELSD method can be used to simultaneously quantify seven saponins, i.e. macranthoidin B, macranthoidin A, dipsacoside B, hederagenin-28-O-β-D-glucopyranosyl(6→1)-O-β-D- glucopyranosyl ester, macranthoside B, macranthoside A, and hederagenin-3-O-α-L-arabinopyranosyl(2→1)-O-α-L-rhamnopyranoside in Flos Lonicerae. Together with the HPLC profiles of iridoids, the HPLC-ELSD profiles of saponins could also be used as an accessory chemical evidence to clarify the botanical origin and comprehensive quality evaluation of Flos Lonicerae.AcknowledgementsThis project is financially supported by Fund for Distinguished Chinese Young Scholars of the National Science Foundation of China (30325046) and the National High Tech Program(2003AA2Z2010).[1]Ministry of Public Health of the People’s Republic of China, Pharmacopoeia ofthe People’s Republic of China, V ol.1, 2000, p. 177.[2]W. Shi, R.B. Shi, Y.R. Lu, Chin. Pharm. J., 34(1999) 724.[3]J.B. Xing, P. Li, D.L. Wen, Chin. Med. Mater., 26(2001) 457.[4]Y.Q. Zhang, L.C. Xu, L.P. Wang, J. Chin. Med. Mater., 21(1996) 204.[5] D. Zhang, Z.W. Li, Y. Jiang, J. Pharm. Anal., 16(1996) 83.[6]T.Z. Wang, Y.M. Li, Huaxiyaoxue Zazhi, 15(2000) 292.[7]J.ZH. Shi, G.T. Liu. Acta Pharm. Sin., 30(1995) 311.[8]Y. P. Liu, J. Liu, X.SH. Jia, et al. Acta Pharmacol. Sin., 13 (1992) 209.[9]Y. P. Liu, J. Liu, X.SH. Jia, et al. Acta Pharmacol. Sin., 13 (1992) 213.[10]J.ZH. Shi, L. Wan, X.F. Chen.ZhongYao YaoLi Yu LinChuang, 6 (1990) 33.[11]J. Liu, L. Xia, X.F. Chen. Acta Pharmacol. Sin., 9 (1988) 395[12]H.J. Li, P. Li, W.C. Ye, J. Chromatogr. A 1008(2003) 167-72.[13]Q. Mao, D. Cao, X.SH. Jia. Acta Pharm. Sin., 28(1993) 273.[14]H. Kizu, S. Hirabayashi, M. Suzuki, et al. Chem. Pharm. Bull., 33(1985) 3473.[15]S. Saito, S. Sumita, N. Tamura, et al. Chem Pharm Bull., 38(1990) 411.[16]Alltech ELSD 2000 Operating Manual, Alltech, 2001, p. 16. In Chinese.[17]J.B. Xing, P. Li, Chin. Med. Mater., 22(1999) 366.Fig. 1 Chemical structures of seven saponins from Lonicera confusa macranthoidin B (1), macranthoidin A (2), dipsacoside B (3), hederagenin-28-O-β-D-glucopyranosyl(6→1)-O-β-D- glucopyranosyl ester (4), macranthoside B (5), macranthoside A (6), and hederagenin-3-O-α-L-arabinopyranosyl(2→1)-O-α-L-rhamnopyranoside (7)Fig. 2Representative HPLC chromatograms of mixed standards and methanol extracts of Flos Lonicerae.Column: Agilent Zorbax SB-C18 column(250 4.6mm, 5.0µm), temperature of 25℃; Detector: ELSD, drift tube temperature 106℃, nitrogen flow-rate 2.6 l/min.A: Mixed standards, B: L. confusa, C: L. japonica, D: L. macranthoides, E: L. hypoglauca, F: L. similes.Table 1 Calibration curves for seven saponinsAnalytes Calibration curve ar2Test range(μg)LOD(μg)LOQ(μg)1 y=6711.9x-377.6 0.9940 0.56–22.01 0.26 0.882 y=7812.6x-411.9 0.9922 0.54–21.63 0.26 0.843 y=6798.5x-299.0 0.9958 0.46–18.42 0.22 0.724 y=12805x-487.9 0.9961 0.38–15.66 0.10 0.345 y=4143.8x-88.62 0.9989 0.42–16.82 0.18 0.246 y=3946.8x-94.4 0.9977 0.40–16.02 0.16 0.207 y=4287.8x-95.2 0.9982 0.42–16.46 0.12 0.22a y: Peak area; x: concentration (mg/ml)Table 2 Reproducibility of the assayAnalyteIntra-day variability Inter-day variability Content (mg/g) Mean RSD (%) Content (mg/g) Mean RSD (%)1 46.1646.2846.2246.22 0.1346.2245.3647.4226.33 2.232 5.385.385.165.31 2.405.285.345.045.22 3.043 4.374.304.184.28 2.244.284.464.024.255.204 nd1)-- -- nd -- --5 1.761.801.821.79 1.701.801.681.841.77 4.706 1.281.241.221.252.451.241.341.201.26 5.727 tr2)-- -- tr -- -- 1): not detected; 2): trace. RSD (%) = (SD/Mean) ×100%Table 3 Recovery of the seven analytesAnalyteOriginal(mg) Spiked(mg)Found(mg)Recovery(%)Mean(%)RSD(%)1 23.0823.1423.1119.7122.8628.1042.7346.1351.0199.7100.699.399.8 0.722.692.672.582.082.913.164.735.515.7698.197.6100.698.8 1.632.172.152.091.732.182.623.884.404.6598.8103.297.799.9 2.94nd1)1.011.050.980.981.101.0297.0104.8104.1102.0 4.250.880.900.910.700.871.081.561.752.0197.197.7101.898.9 2.660.640.620.610.450.610.751.081.211.3397.796.796.096.8 0.97tr2)1.021.101.081.031.111.07100.9102.799.1100.9 1.81): not detected; 2): trace.a Recovery (%) = (Amount found –Original amount)/ Amount spiked ×100%, RSD (%) = (SD/Mean) ×100%Table 4 Contents of seven saponins in Lonicera spp.Content (mg/g)1 2 3 4 5 6 7 L. confusa45.65±0.32 5.13±0.08 4.45±0.11tr1) 2.04±0.04tr 1.81±0.03 L. japonica nd2)nd 3.44±0.09nd nd nd nd L. macranthoides46.22±0.06 5.31±0.13 4.28±0.10 tr 1.79±0.03 1.25±0.03 tr L. hypoglauca11.17±0.07 nq3)53.78±1.18nd 1.72±0.02 2.23±0.06 2.52±0.04 L. similes41.22±0.25 4.57±0.07 3.79±0.09nd 1.75±0.02tr nd 1): trace; 2): not detected.. 3) not quantified owing to the suspicious purity of the peak.。
常用蛋白酶切割位点
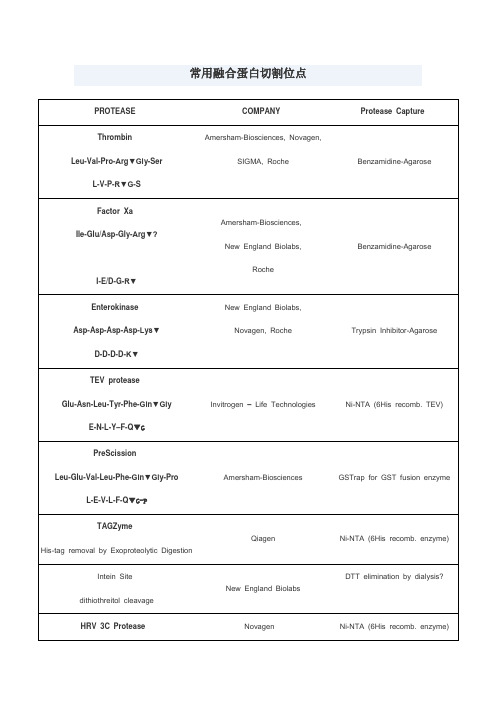
4.溴化氰处理,专一性的切割甲硫氨酸羧基端的肽键。
SIGMA, Roche
Benzamidine-Agarose
Factor Xa
Ile-Glu/Asp-Gly-Arg▼?
I-E/D-G-R▼
Amersham-Biosciences,
New England Biolabs,
Roche
Benzamidine-Agarose
Enterokinase
Asp-Asp-Asp-Asp-Lys▼
羧肽酶
羧肽酶B可以切割C端的Lys或Arg;羧肽酶A可以切割C端除了Lys、Arg、Pro的氨基酸,但如果倒数第二个氨基酸为Pro两种羧肽酶均不能作用
1.胰蛋白酶属肽链内切酶,能把多肽链中Lys和Arg残基中的羧基侧切断。
2.胰凝乳蛋白酶(亦称糜蛋白酶)属肽链内切酶,主要切断多肽链中的芳香族氨基酸(Phe、Trp、Tyr)残基的羧基一侧。
LifeSensors
Ni-NTA (6His recomb. enzyme)
Kex-2
-Arg-X-Lys/Arg-Arg▼
Invitrogen – Life Technologies,
Ni-NTA (6His recomb. enzyme)
KEX2对arg的专一性高,要求最重要。
Arg前为lys效率最高,不切-Arg-lys,Pro影响KEX2切割
Ni-NTA (6His recomb. TEV)
PreScission
Leu-Glu-Val-Leu-Phe-Gln▼Gly-Pro
稳定性英文版

HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationFLUOXETINE HClC17H18F3NO•HClM.W. = 345.79CAS — 59333-67-4STABILITY INDICATINGA S S A Y V A L I D A T I O NMethod is suitable for:ýIn-process controlþProduct ReleaseþStability indicating analysis (Suitability - US/EU Product) CAUTIONFLUOXETINE HYDROCHLORIDE IS A HAZARDOUS CHEMICAL AND SHOULD BE HANDLED ONLY UNDER CONDITIONS SUITABLE FOR HAZARDOUS WORK.IT IS HIGHLY PRESSURE SENSITIVE AND ADEQUATE PRECAUTIONS SHOULD BE TAKEN TO AVOID ANY MECHANICAL FORCE (SUCH AS GRINDING, CRUSHING, ETC.) ON THE POWDER.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationTABLE OF CONTENTS INTRODUCTION........................................................................................................................ PRECISION............................................................................................................................... System Repeatability ................................................................................................................ Method Repeatability................................................................................................................. Intermediate Precision .............................................................................................................. LINEARITY................................................................................................................................ RANGE...................................................................................................................................... ACCURACY............................................................................................................................... Accuracy of Standard Injections................................................................................................ Accuracy of the Drug Product.................................................................................................... VALIDATION OF FLUOXETINE HCl AT LOW CONCENTRATION........................................... Linearity at Low Concentrations................................................................................................. Accuracy of Fluoxetine HCl at Low Concentration..................................................................... System Repeatability................................................................................................................. Quantitation Limit....................................................................................................................... Detection Limit........................................................................................................................... VALIDATION FOR META-FLUOXETINE HCl (POSSIBLE IMPURITIES).................................. Meta-Fluoxetine HCl linearity at 0.05% - 1.0%........................................................................... Detection Limit for Fluoxetine HCl.............................................................................................. Quantitation Limit for Meta Fluoxetine HCl................................................................................ Accuracy for Meta-Fluoxetine HCl ............................................................................................ Method Repeatability for Meta-Fluoxetine HCl........................................................................... Intermediate Precision for Meta-Fluoxetine HCl......................................................................... SPECIFICITY - STABILITY INDICATING EVALUATION OF THE METHOD............................. FORCED DEGRADATION OF FINISHED PRODUCT AND STANDARD..................................1. Unstressed analysis...............................................................................................................2. Acid Hydrolysis stressed analysis..........................................................................................3. Base hydrolysis stressed analysis.........................................................................................4. Oxidation stressed analysis...................................................................................................5. Sunlight stressed analysis.....................................................................................................6. Heat of solution stressed analysis.........................................................................................7. Heat of powder stressed analysis.......................................................................................... System Suitability stressed analysis.......................................................................................... Placebo...................................................................................................................................... STABILITY OF STANDARD AND SAMPLE SOLUTIONS......................................................... Standard Solution...................................................................................................................... Sample Solutions....................................................................................................................... ROBUSTNESS.......................................................................................................................... Extraction................................................................................................................................... Factorial Design......................................................................................................................... CONCLUSION...........................................................................................................................ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationBACKGROUNDTherapeutically, Fluoxetine hydrochloride is a classified as a selective serotonin-reuptake inhibitor. Effectively used for the treatment of various depressions. Fluoxetine hydrochloride has been shown to have comparable efficacy to tricyclic antidepressants but with fewer anticholinergic side effects. The patent expiry becomes effective in 2001 (US). INTRODUCTIONFluoxetine capsules were prepared in two dosage strengths: 10mg and 20mg dosage strengths with the same capsule weight. The formulas are essentially similar and geometrically equivalent with the same ingredients and proportions. Minor changes in non-active proportions account for the change in active ingredient amounts from the 10 and 20 mg strength.The following validation, for the method SI-IAG-206-02 , includes assay and determination of Meta-Fluoxetine by HPLC, is based on the analytical method validation SI-IAG-209-06. Currently the method is the in-house method performed for Stability Studies. The Validation was performed on the 20mg dosage samples, IAG-21-001 and IAG-21-002.In the forced degradation studies, the two placebo samples were also used. PRECISIONSYSTEM REPEATABILITYFive replicate injections of the standard solution at the concentration of 0.4242mg/mL as described in method SI-IAG-206-02 were made and the relative standard deviation (RSD) of the peak areas was calculated.SAMPLE PEAK AREA#15390#25406#35405#45405#55406Average5402.7SD 6.1% RSD0.1ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::PRECISION - Method RepeatabilityThe full HPLC method as described in SI-IAG-206-02 was carried-out on the finished product IAG-21-001 for the 20mg dosage form. The method repeated six times and the relative standard deviation (RSD) was calculated.SAMPLENumber%ASSAYof labeled amountI 96.9II 97.8III 98.2IV 97.4V 97.7VI 98.5(%) Average97.7SD 0.6(%) RSD0.6PRECISION - Intermediate PrecisionThe full method as described in SI-IAG-206-02 was carried-out on the finished product IAG-21-001 for the 20mg dosage form. The method was repeated six times by a second analyst on a different day using a different HPLC instrument. The average assay and the relative standard deviation (RSD) were calculated.SAMPLENumber% ASSAYof labeled amountI 98.3II 96.3III 94.6IV 96.3V 97.8VI 93.3Average (%)96.1SD 2.0RSD (%)2.1The difference between the average results of method repeatability and the intermediate precision is 1.7%.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationLINEARITYStandard solutions were prepared at 50% to 200% of the nominal concentration required by the assay procedure. Linear regression analysis demonstrated acceptability of the method for quantitative analysis over the concentration range required. Y-Intercept was found to be insignificant.RANGEDifferent concentrations of the sample (IAG-21-001) for the 20mg dosage form were prepared, covering between 50% - 200% of the nominal weight of the sample.Conc. (%)Conc. (mg/mL)Peak Area% Assayof labeled amount500.20116235096.7700.27935334099.21000.39734463296.61500.64480757797.52000.79448939497.9(%) Average97.6SD 1.0(%) RSD 1.0ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::RANGE (cont.)The results demonstrate linearity as well over the specified range.Correlation coefficient (RSQ)0.99981 Slope11808.3Y -Interceptresponse at 100%* 100 (%) 0.3%ACCURACYACCURACY OF STANDARD INJECTIONSFive (5) replicate injections of the working standard solution at concentration of 0.4242mg/mL, as described in method SI-IAG-206-02 were made.INJECTIONNO.PEAK AREA%ACCURACYI 539299.7II 540599.9III 540499.9IV 5406100.0V 5407100.0Average 5402.899.9%SD 6.10.1RSD, (%)0.10.1The percent deviation from the true value wasdetermined from the linear regression lineHPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::ACCURACY OF THE DRUG PRODUCTAdmixtures of non-actives (placebo, batch IAG-21-001 ) with Fluoxetine HCl were prepared at the same proportion as in a capsule (70%-180% of the nominal concentration).Three preparations were made for each concentration and the recovery was calculated.Conc.(%)Placebo Wt.(mg)Fluoxetine HCl Wt.(mg)Peak Area%Accuracy Average (%)70%7079.477.843465102.27079.687.873427100.77079.618.013465100.0101.0100%10079.6211.25476397.910080.8011.42491799.610079.6011.42485498.398.6130%13079.7214.90640599.413080.3114.75632899.213081.3314.766402100.399.618079.9920.10863699.318079.3820.45879499.418080.0820.32874899.599.4Placebo, Batch Lot IAG-21-001HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::VALIDATION OF FLUOXETINE HClAT LOW CONCENTRATIONLINEARITY AT LOW CONCENTRATIONSStandard solution of Fluoxetine were prepared at approximately 0.02%-1.0% of the working concentration required by the method SI-IAG-206-02. Linear regression analysis demonstrated acceptability of the method for quantitative analysis over this range.ACCURACY OF FLUOXETINE HCl AT LOW CONCENTRATIONThe peak areas of the standard solution at the working concentration were measured and the percent deviation from the true value, as determined from the linear regression was calculated.SAMPLECONC.µg/100mLAREA FOUND%ACCURACYI 470.56258499.7II 470.56359098.1III 470.561585101.3IV 470.561940100.7V 470.56252599.8VI 470.56271599.5(%) AverageSlope = 132.7395299.9SD Y-Intercept = -65.872371.1(%) RSD1.1HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSystem RepeatabilitySix replicate injections of standard solution at 0.02% and 0.05% of working concentration as described in method SI-IAG-206-02 were made and the relative standard deviation was calculated.SAMPLE FLUOXETINE HCl AREA0.02%0.05%I10173623II11503731III10103475IV10623390V10393315VI10953235Average10623462RSD, (%) 5.0 5.4Quantitation Limit - QLThe quantitation limit ( QL) was established by determining the minimum level at which the analyte was quantified. The quantitation limit for Fluoxetine HCl is 0.02% of the working standard concentration with resulting RSD (for six injections) of 5.0%. Detection Limit - DLThe detection limit (DL) was established by determining the minimum level at which the analyte was reliably detected. The detection limit of Fluoxetine HCl is about 0.01% of the working standard concentration.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::VALIDATION FOR META-FLUOXETINE HCl(EVALUATING POSSIBLE IMPURITIES)Meta-Fluoxetine HCl linearity at 0.05% - 1.0%Relative Response Factor (F)Relative response factor for Meta-Fluoxetine HCl was determined as slope of Fluoxetine HCl divided by the slope of Meta-Fluoxetine HCl from the linearity graphs (analysed at the same time).F =132.7395274.859534= 1.8Detection Limit (DL) for Fluoxetine HClThe detection limit (DL) was established by determining the minimum level at which the analyte was reliably detected.Detection limit for Meta Fluoxetine HCl is about 0.02%.Quantitation Limit (QL) for Meta-Fluoxetine HClThe QL is determined by the analysis of samples with known concentration of Meta-Fluoxetine HCl and by establishing the minimum level at which the Meta-Fluoxetine HCl can be quantified with acceptable accuracy and precision.Six individual preparations of standard and placebo spiked with Meta-Fluoxetine HCl solution to give solution with 0.05% of Meta Fluoxetine HCl, were injected into the HPLC and the recovery was calculated.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::META-FLUOXETINE HCl[RECOVERY IN SPIKED SAMPLES].Approx.Conc.(%)Known Conc.(µg/100ml)Area in SpikedSampleFound Conc.(µg/100mL)Recovery (%)0.0521.783326125.735118.10.0521.783326825.821118.50.0521.783292021.55799.00.0521.783324125.490117.00.0521.783287220.96996.30.0521.783328526.030119.5(%) AVERAGE111.4SD The recovery result of 6 samples is between 80%-120%.10.7(%) RSDQL for Meta Fluoxetine HCl is 0.05%.9.6Accuracy for Meta Fluoxetine HClDetermination of Accuracy for Meta-Fluoxetine HCl impurity was assessed using triplicate samples (of the drug product) spiked with known quantities of Meta Fluoxetine HCl impurity at three concentrations levels (namely 80%, 100% and 120% of the specified limit - 0.05%).The results are within specifications:For 0.4% and 0.5% recovery of 85% -115%For 0.6% recovery of 90%-110%HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::META-FLUOXETINE HCl[RECOVERY IN SPIKED SAMPLES]Approx.Conc.(%)Known Conc.(µg/100mL)Area in spikedSample Found Conc.(µg/100mL)Recovery (%)[0.4%]0.4174.2614283182.66104.820.4174.2614606187.11107.370.4174.2614351183.59105.36[0.5%]0.5217.8317344224.85103.220.5217.8316713216.1599.230.5217.8317341224.81103.20[0.6%]0.6261.3918367238.9591.420.6261.3920606269.81103.220.6261.3920237264.73101.28RECOVERY DATA DETERMINED IN SPIKED SAMPLESHPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::REPEATABILITYMethod Repeatability - Meta Fluoxetine HClThe full method (as described in SI-IAG-206-02) was carried out on the finished drug product representing lot number IAG-21-001-(1). The HPLC method repeated serially, six times and the relative standard deviation (RSD) was calculated.IAG-21-001 20mg CAPSULES - FLUOXETINESample% Meta Fluoxetine % Meta-Fluoxetine 1 in Spiked Solution10.0260.09520.0270.08630.0320.07740.0300.07450.0240.09060.0280.063AVERAGE (%)0.0280.081SD 0.0030.012RSD, (%)10.314.51NOTE :All results are less than QL (0.05%) therefore spiked samples with 0.05% Meta Fluoxetine HCl were injected.HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationED. N0: 04Effective Date:APPROVED::Intermediate Precision - Meta-Fluoxetine HClThe full method as described in SI-IAG-206-02 was applied on the finished product IAG-21-001-(1) .It was repeated six times, with a different analyst on a different day using a different HPLC instrument.The difference between the average results obtained by the method repeatability and the intermediate precision was less than 30.0%, (11.4% for Meta-Fluoxetine HCl as is and 28.5% for spiked solution).IAG-21-001 20mg - CAPSULES FLUOXETINESample N o:Percentage Meta-fluoxetine% Meta-fluoxetine 1 in spiked solution10.0260.06920.0270.05730.0120.06140.0210.05850.0360.05560.0270.079(%) AVERAGE0.0250.063SD 0.0080.009(%) RSD31.514.51NOTE:All results obtained were well below the QL (0.05%) thus spiked samples slightly greater than 0.05% Meta-Fluoxetine HCl were injected. The RSD at the QL of the spiked solution was 14.5%HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSPECIFICITY - STABILITY INDICATING EVALUATIONDemonstration of the Stability Indicating parameters of the HPLC assay method [SI-IAG-206-02] for Fluoxetine 10 & 20mg capsules, a suitable photo-diode array detector was incorporated utilizing a commercial chromatography software managing system2, and applied to analyze a range of stressed samples of the finished drug product.GLOSSARY of PEAK PURITY RESULT NOTATION (as reported2):Purity Angle-is a measure of spectral non-homogeneity across a peak, i.e. the weighed average of all spectral contrast angles calculated by comparing all spectra in the integrated peak against the peak apex spectrum.Purity Threshold-is the sum of noise angle3 and solvent angle4. It is the limit of detection of shape differences between two spectra.Match Angle-is a comparison of the spectrum at the peak apex against a library spectrum.Match Threshold-is the sum of the match noise angle3 and match solvent angle4.3Noise Angle-is a measure of spectral non-homogeneity caused by system noise.4Solvent Angle-is a measure of spectral non-homogeneity caused by solvent composition.OVERVIEWT he assay of the main peak in each stressed solution is calculated according to the assay method SI-IAG-206-02, against the Standard Solution, injected on the same day.I f the Purity Angle is smaller than the Purity Threshold and the Match Angle is smaller than the Match Threshold, no significant differences between spectra can be detected. As a result no spectroscopic evidence for co-elution is evident and the peak is considered to be pure.T he stressed condition study indicated that the Fluoxetine peak is free from any appreciable degradation interference under the stressed conditions tested. Observed degradation products peaks were well separated from the main peak.1® PDA-996 Waters™ ; 2[Millennium 2010]ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationFORCED DEGRADATION OF FINISHED PRODUCT & STANDARD 1.UNSTRESSED SAMPLE1.1.Sample IAG-21-001 (2) (20mg/capsule) was prepared as stated in SI-IAG-206-02 and injected into the HPLC system. The calculated assay is 98.5%.SAMPLE - UNSTRESSEDFluoxetine:Purity Angle:0.075Match Angle:0.407Purity Threshold:0.142Match Threshold:0.4251.2.Standard solution was prepared as stated in method SI-IAG-206-02 and injected into the HPLC system. The calculated assay is 100.0%.Fluoxetine:Purity Angle:0.078Match Angle:0.379Purity Threshold:0.146Match Threshold:0.4272.ACID HYDROLYSIS2.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as in method SI-IAG-206-02 : An amount equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent was added and the solution sonicated for 10 minutes. 1mL of conc. HCl was added to this solution The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with NaOH 10N, made up to volume with Diluent and injected into the HPLC system after filtration.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 98.8%.SAMPLE- ACID HYDROLYSISFluoxetine peak:Purity Angle:0.055Match Angle:0.143Purity Threshold:0.096Match Threshold:0.3712.2.Standard solution was prepared as in method SI-IAG-206-02 : about 22mg Fluoxetine HCl were weighed into a 50mL volumetric flask. 20mL Diluent were added. 2mL of conc. HCl were added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with NaOH 10N, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 97.2%.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSTANDARD - ACID HYDROLYSISFluoxetine peak:Purity Angle:0.060Match Angle:0.060Purity Threshold:0.099Match Threshold:0.3713.BASE HYDROLYSIS3.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as per method SI-IAG-206-02 : An amount equivalent to 20mg Fluoxetine was weight into a 50mL volumetric flask. 20mL Diluent was added and the solution sonicated for 10 minutes. 1mL of 5N NaOH was added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH = 5.5 with 5N HCl, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease. Assay result obtained - 99.3%.SAMPLE - BASE HYDROLYSISFluoxetine peak:Purity Angle:0.063Match Angle:0.065Purity Threshold:0.099Match Threshold:0.3623.2.Standard stock solution was prepared as per method SI-IAG-206-02 : About 22mg Fluoxetine HCl was weighed into a 50mL volumetric flask. 20mL Diluent was added. 2mL of 5N NaOH was added to this solution. The solution was allowed to stand for 18 hours, then adjusted to about pH=5.5 with 5N HCl, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity did NOT decrease - 99.5%.STANDARD - BASE HYDROLYSISFluoxetine peak:Purity Angle:0.081Match Angle:0.096Purity Threshold:0.103Match Threshold:0.3634.OXIDATION4.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as per method SI-IAG-206-02. An equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent added and the solution sonicated for 10 minutes.1.0mL of 30% H2O2 was added to the solution and allowed to stand for 5 hours, then made up to volume with Diluent, filtered and injected into HPLC system.Fluoxetine peak intensity decreased to 95.2%.ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSAMPLE - OXIDATIONFluoxetine peak:Purity Angle:0.090Match Angle:0.400Purity Threshold:0.154Match Threshold:0.4294.2.Standard solution was prepared as in method SI-IAG-206-02 : about 22mg Fluoxetine HCl were weighed into a 50mL volumetric flask and 25mL Diluent were added. 2mL of 30% H2O2 were added to this solution which was standing for 5 hours, made up to volume with Diluent and injected into the HPLC system.Fluoxetine peak intensity decreased to 95.8%.STANDARD - OXIDATIONFluoxetine peak:Purity Angle:0.083Match Angle:0.416Purity Threshold:0.153Match Threshold:0.4295.SUNLIGHT5.1.Sample solution of IAG-21-001 (2) (20mg/capsule) was prepared as in method SI-IAG-206-02 . The solution was exposed to 500w/hr. cell sunlight for 1hour. The BST was set to 35°C and the ACT was 45°C. The vials were placed in a horizontal position (4mm vials, National + Septum were used). A Dark control solution was tested. A 2%w/v quinine solution was used as the reference absorbance solution.Fluoxetine peak decreased to 91.2% and the dark control solution showed assay of 97.0%. The difference in the absorbance in the quinine solution is 0.4227AU.Additional peak was observed at RRT of 1.5 (2.7%).The total percent of Fluoxetine peak with the degradation peak is about 93.9%.SAMPLE - SUNLIGHTFluoxetine peak:Purity Angle:0.093Match Angle:0.583Purity Threshold:0.148Match Threshold:0.825 ED. N0: 04Effective Date:APPROVED::HPLC ASSAY with DETERMINATION OF META-FLUOXETINE HCl.ANALYTICAL METHOD VALIDATION10 and 20mg Fluoxetine Capsules HPLC DeterminationSUNLIGHT (Cont.)5.2.Working standard solution was prepared as in method SI-IAG-206-02 . The solution was exposed to 500w/hr. cell sunlight for 1.5 hour. The BST was set to 35°C and the ACT was 42°C. The vials were placed in a horizontal position (4mm vials, National + Septum were used). A Dark control solution was tested. A 2%w/v quinine solution was used as the reference absorbance solution.Fluoxetine peak was decreased to 95.2% and the dark control solution showed assay of 99.5%.The difference in the absorbance in the quinine solution is 0.4227AU.Additional peak were observed at RRT of 1.5 (2.3).The total percent of Fluoxetine peak with the degradation peak is about 97.5%. STANDARD - SUNLIGHTFluoxetine peak:Purity Angle:0.067Match Angle:0.389Purity Threshold:0.134Match Threshold:0.8196.HEAT OF SOLUTION6.1.Sample solution of IAG-21-001-(2) (20 mg/capsule) was prepared as in method SI-IAG-206-02 . Equivalent to 20mg Fluoxetine was weighed into a 50mL volumetric flask. 20mL Diluent was added and the solution was sonicated for 10 minutes and made up to volume with Diluent. 4mL solution was transferred into a suitable crucible, heated at 105°C in an oven for 2 hours. The sample was cooled to ambient temperature, filtered and injected into the HPLC system.Fluoxetine peak was decreased to 93.3%.SAMPLE - HEAT OF SOLUTION [105o C]Fluoxetine peak:Purity Angle:0.062Match Angle:0.460Purity Threshold:0.131Match Threshold:0.8186.2.Standard Working Solution (WS) was prepared under method SI-IAG-206-02 . 4mL of the working solution was transferred into a suitable crucible, placed in an oven at 105°C for 2 hours, cooled to ambient temperature and injected into the HPLC system.Fluoxetine peak intensity did not decrease - 100.5%.ED. N0: 04Effective Date:APPROVED::。
百普乐(培哚普利吲达帕胺片)

特殊警告 与培哚普利相关:
● 在免疫功能低下患者发生中性白细胞减少症 / 粒细胞缺乏症的危险 中性粒细胞减少症的危险与剂量及患者类型相关,并取决于患者的临床情况。没有并发症的患者极少会出现这种情况,但是与胶原血管性疾病相关的肾 功能不全的患者可能发生,如系统性红斑狼疮或硬皮病患者以及使用免疫抑制剂治疗的患者。 停止使用血管紧张素转化酶抑制剂治疗,危险性可消失。 严格遵守预先规定的剂量用药可能是防止事件发生的最好办法。但是,如果这些患者需要使用血管紧张素转化酶抑制剂,应慎重评估风险 / 效益比值。
儿童 百普乐不能用于儿童,因为儿童单独应用或联合应用培哚普利的疗效和耐受性尚未确定。
[不良反应]
服用培哚普利可抑制肾素-血管紧张素-醛固酮轴而使吲达帕胺所致的失钾减少。服用百普乐的 2%患者出现低钾血症(钾离子水平< 3.4mmol/l)。
胃肠道 - 通常发生(> 1/100, < 1/10):便秘、口干、恶心、上腹痛、厌食、腹痛、味觉障碍。 - 极少发生(< 1/10, 000):胰腺炎。 - 在肝功能不全病例中,有引发肝性脑病的可能性(见禁忌和注意事项)。
一个月内血压即出现下降,无急速抗药反应;停药后无反弹作用。在临床试验中,同时给予培哚普利和吲达帕胺,与分别单独使用这二种药物相比,可 产生具有协同作用的抗高血压疗效。
与培哚普利相关: 培哚普利可以治疗各种程度的高血压:轻度到中度或重度。可以降低卧位和立位的收缩压和舒张压。 最大降压作用出现在服用单一剂量后 4-6 小时,降压作用可持续 24 小时以上。
培哚普利在低或正常肾素水平的患者中也可产生抗高血压作用。
培哚普利通过它的活性代谢产物—培哚普利拉产生作用。其它的代谢产物均无活性。
培哚普利可减轻心脏负荷: - 通过改变前列腺素代谢产生扩张静脉的作用:减轻前负荷, - 通过降低总外周血管阻力:减轻后负荷。