高等有机第二章-溶剂化效应
高等有机化学习题

高等有机化学习题第一章 化学键一、用共振轮说明下列问题1) 联本中的C 1-C 2键长为什么比乙烷中的键长短?联苯的硝化反应为什么主要发生在2-位和4-位?联苯的共振结构式可表是如下:(1) 由共振结构式可以看出C 1-C 2键有双键结构的贡献,故比乙烷的C 1-C 2键短。
(2) 由共振结构式可以看出邻对位负电荷相对集中,故有利于发生硝化反应。
2) 方酸为什么是强酸?(强于硫酸) 方酸的共振结构式可表是如下:对吗?由方酸的共振结构式可以看出方酸的电子离域效果更好。
二、试推测6,6-二苯基富烯的亲电取代发生于哪个环,哪个位置?亲核取代发生于哪个环,哪个位置?6,6-二苯基富烯的共振式如下:由6,6-二苯基富烯的共振式可以看出,亲电取代发生在五元环的2位上,而亲核取代发生在苯环的2位上。
三、计算烯丙基正离子和环丙烯正离子π分子轨道的总能量,并比较两者的稳定性。
烯丙基正离子有两个电子在成键轨道上其总能量为 E 烯丙基正离子=2E 1=2(α+1.414β)=2α+2.828β11'O HO O O O OH O O O OH O O OHO O O O S O O HO O S O O OH O S O O O S OOOH环丙烯正离子有两个电子在成键轨道上其总能量为 E 环丙烯正离子=2E 1=2(α+2β)=2α+2β 能量差值为E 烯丙基正离子- E 环丙烯正离子=(2α+2.828β)- (2α+2β)=0.828β 因此,环丙烯正离子比烯丙基正离子稳定。
四、用HMO 法分别说明烯丙基负离子和环丙烯负离子的电子排布和成键情况,并比较两者稳定性。
五、简要说明1)吡咯和吡啶分子的极性方向相反,为什么?吡咯分子中氮原子给出一对为共用电子参与了共轭分子的大π键,也就是电子从氮原子流向五员环,而吡啶分子中氮原子只拿出一个电子参与共轭,并且氮原子的电负性大于碳原子使电子流向氮原子的方向。
因此,两个分子的极性正好相反。
有机反应中溶剂效应对亲核反应的影响

使该类落 荆有 较强极性
,
容 易 使 亲核
,
对 反 应 的 加邃作 用越 强
而且
该类溶荆 的负 端 位 阻 小
容易 使 正
不 能 使丫
例如
一
于 〔 砂 妙… 一
一
’
…
‘
离子
二
专 一 性溶 剂 化 而溶荆 的 正 端 由于 位 阻 大
,
,
一 第 二 种 类 型 对 于 从起 始 反 应 物 变 为 活 化 配 合 物时异号
、
,
,
力 而 专 一 性 力 只 在 一 定 分 子 结 构之 间 才 能 发 生
,
,
、
落 荆的分类 溶 剂 按 其 起 氢 键给 体作 用 分 类 溶 剂 按 照 其 起 氢 健给 体
的 能 力 可 以 分 为质 子 传 递 型 溶剂 和 非质 子 传 递 型 溶剂 两 大
分 类 以 及 溶 剂 和溶 质 之 间 的 相 互 作 用
综 上 所 述 笔 者讨 论 了有 机 反 应 中溶 荆效应对 于 亲核取
,
使 反 应质点 丫专 一 性 溶 剂 化
从 而 降 低丫 的反应活性 和 该类
,
代 反应的 影 响做 了分析 希 望 这 方 面 的 内容 对 于 在亲核取 代
反 应中 正 确 选择 有 机 溶 剂 能 起到一 定 的辅助 作 用
和 专 一性 力 其 中库 仑 力 和 范 德 华 力 是 普 追 存 在 的 非专 一 性
中 主 反 应 和 副 反 应 有 竞 争 如 果 选 择 合 适的 溶 剂就 可 以 使
主 反 应 显 著 加速 并 且 能 有效 地 抑 制 副反 应 另外 溶剂 还 会 形 响 反 应 历 程 反应方 向 和立 体 化 学 因此 了解溶剂 的 性 质
溶剂效应介绍 PPT

溶剂
乙醇
甲醇 甲酸
水
介电常数
24.55
32.7
58.5 78.39
相对反应速度 1
9
12200 33500
上述静电溶剂化理论是一个简单的定性的纯静电理论,
有一定的局限性。它忽略反应中的熵变以及溶剂与溶剂的 相互作用等等。因此,有些情况例外。
7.3 特殊溶剂化效应
特殊溶剂化可分为负离子的特殊溶剂化和正离子的
渡态与溶剂分子间的静电作用,以比较起始反应物和过
渡态电荷分离程度的大小,从而可以预测溶剂极性对离
子型反应速率的影响。对过渡态比起始物分子具有较大
电荷分离程度的反应,溶剂极性的增加使反应速率加快;
而对于过渡态比起始反应物分子电荷分离程度减少的反
应,溶剂极性的增加使反应速率减慢。对溶剂极性减少
的情况来说,则情况刚好是相反的。能量变化与溶剂极
7 溶剂效应
大多数有机反应是在溶剂中进行的。很早以前人们 就知道溶剂对化学反应有重要的影晌。但作为一个溶剂 理论,却是在60年代才发展起来的,比电子理论的发展 慢得多。最近一.二十年运用近代科学技术对溶剂效应 展开了系统的和广泛的研究,得到了一些有关溶剂化的 规律和理论,它们对进一步深入认识化学反应具有重要 的指导意义。例如,1983年有人报导了OH—对CH3Br的亲 核取代反应,观察了溶剂分子H2O的数目对OH-亲核性的 影响。发现一个赤裸的OH—的亲核性比它在水溶液中大 16个数量级[即使有一个水分子在旁也能使它的亲核活 性减少几十倍。
5
大家好
R L S R L s R ‖ L s R s L s
溶质
紧密离 子对
(A)
溶剂分离 离子对
(B)
溶剂 溶剂 化正 化负 离子 离子
有机化学中的溶剂化效应——溶剂对反应历程和立体化学的影响

有机化学中的溶剂化效应——溶剂对反应历程和立体化学的影响作者:蒙慧芹来源:《赤峰学院学报·自然科学版》 2011年第6期蒙慧芹(赤峰学院化学化工学院,内蒙古赤峰 024000)摘要:有机化学反应大多数是在溶剂中进行的,溶剂对化学反应起着非常重要的作用.在已知的300多种溶剂和无数混合溶剂中,选择适当溶剂作为反应场所与选择合适的催化剂相比,具有更重要的意义.本文主要介绍了溶剂的分类和极性,有选择地讨论了某些反应,当改变溶剂时,将引起这些反应在反应历程和立体化学方面的变化.关键词:溶剂;反应历程;立体化学;影响中图分类号:O621文献标识码:A文章编号:1673-260X(2011)06-0001-03许多化学反应是在溶剂中进行的,溶剂在有机化学中的应用十分普遍,它不仅影响着反应平衡及反应速率,而且对反应历程和立体化学也有着重大影响.那么溶剂具有怎样的分类,以及决定溶剂极性的参数有那些?它们对有机反应历程和立体化学有什么影响?分别讨论如下:1关于溶剂的分类和极性1.1溶剂的分类溶剂的数量繁多,有各种对溶剂进行分类的方法,通常从以下四个方面对溶剂加以分类:1.1.1按照溶剂的化学结构把溶剂分为烃类、卤代烃类、醇类、酯类、胺类等等.1.1.2按照溶剂的酸碱性把溶剂分为酸性溶剂、碱性溶剂、中性溶剂等等.1.1.3按照溶剂的物理性质如:根据沸点、相对密度、粘度、极性等分类.1.1.4按照溶剂与溶质的专属作用把溶剂分类.专属作用是指溶剂与溶质间的相互作用,包括氢键、电子授受、电荷转移等.后两种分类体系较为科学,更能反映其实质,是目前常用的分类方法.若将它们结合起来,可把溶剂分为以下四类:极性溶剂、非极性溶剂、质子型溶剂和非质子型溶剂.1.2溶剂的极性溶剂的极性有很多量度方法,长期以来,溶剂极性的大小,常以偶极矩值(μ)和介电常数(ε)来量度.有机溶剂的永久偶极矩处于0到18.3×10-30c·m(0到5、5D)之间.从烃类溶剂到含有极性基团的溶剂,其偶极矩值逐步提高.有机溶剂的介电常数一般为2~190.通常认为ε>15,偶极矩大于8.34×10-30c·m(2、5D)为极性溶剂,若ε<15,偶极矩处于0~6.67-30c·m(0~2D)的溶剂为非极性溶剂.表1列出了一些溶剂的介电常数.2溶剂对反应历程和立体化学的影响2.1溶剂对亲核取代反应的影响饱和碳原子上的亲核取代反应有两种典型的历程:单分子亲核取代反应(SN1)历程和双分子亲核取代反应(SN2)历程.SN1反应历程:SN1反应是分两步进行的,第一步是控制反应速率的一步,即慢的一步.在这一步中极性较小的底物分子逐渐发生化学键的解离,形成极性较大的过渡态,最后离解成碳正离子,即在反应过程中极性加大,增加溶剂的极性有利于过渡态的形成.溶剂与分子或离子通过静电力而结合的作用叫做溶剂化作用.溶剂与SN1反应的过渡态有偶极-偶极相互作用,即溶剂化作用.底物在形成过渡态时需要能量,此能量可由溶剂与过渡态偶极-偶极相互作用时所释放的能量提供,因此溶剂的极性大,溶剂化的力量也大,提供的能量也大,离解就能很快进行.SN2反应历程:SN2反应是同步进行的协同反应,溶剂对SN2反应历程的影响比较复杂.当正离子与中性试剂、中性底物与负离子、正离子与负离子反应时,由于生成的过渡态中没有离子存在,电荷得到了分散和消失.因此增加溶剂极性,不利于过渡态的形成,使反应速率降低.反之,当中性底物与中性试剂反应时,生成的过渡态反而比底物的电荷增加.因此,增加溶剂极性,有利于过渡态的形成,使反应速率增加.如下列为中性底物与负离子的反应历程:在形成过渡态时,由电荷比较集中的亲核试剂变成电荷比较分散的过渡态,亲核试剂的一部分负电荷通过R传给了L,过渡态的负电荷比较分散,不如亲核试剂集中,因而过渡态的极性不如亲核试剂大,增加溶剂的极性,使极性大的亲核试剂溶剂化,而不利于SN2过渡态的形成.在反应a中,溶剂乙醚的极性低,有利于反应按SN2历程进行,所以亲核试剂碘离子向位阻小的甲基进攻,生成了碘甲烷和叔丁醇在反应b中,溶剂水的极性高,有利于反应按SN1历程进行,因而生成了甲醇和叔丁基碘:2.2溶剂对亲电加成反应的影响烯烃的亲电加成也有两种典型的历程,即碳正离子中间体历程和环状鎓离子历程.碳正离子历程:在CH3NO2溶剂中,反式加成:顺式加成,1:9在CCI4溶剂中,反式加成:顺式加成,10:1在强极性的溶剂硝基甲烷(介电常数为36)中,碳正离子由于较强的溶剂化作用而比溴鎓离子稳定,因而以顺式加成产物为主.在弱极性的溶剂四氯化碳(介电常数为2.2)中,由于溶剂化作用微弱,所以溴鎓离子比碳正离子稳定,于是得到反式加成为主的产物.炔烃与卤素的亲电加成类似与烯烃,在不同溶剂中进行炔烃的亲电加成反应时,加成的立体化学也不同.例如,苯乙炔与溴的加成在不同的溶剂中生成的产物比例如下:实例3[3]2.3溶剂对环加成反应的影响在光化学诱导的[2+2]环加成反应中,立体化学也受溶剂极性的影响.实例4[4]当溶剂的极性提高时,反向/顺向产物的比例随之降低,这是由于溶剂的极性愈强,将愈强烈地使活化配合物溶剂化,导致偶极性的顺向产物的增加.2.4溶剂对Wittig反应的影响在Wittig反应中,生成的烯烃如有顺、反异构体时,溶剂对所产生异构体的比例也有很大影响.经研究证明:反应在极性溶剂中进行,有利于顺式异构体的生成;在非极性溶剂中进行,则有利于反式异构体的生成.这个原理已在昆虫激素的生产实践上得到成功的应用.例如:组成.当反式比例占10%左右时,诱蛾活性最高.过去,按乙炔路线合成时,需先分别合成顺、反两种异构体,再按一定比例混合,路线长,手续麻烦.中国科学院、北京动物研究所用Wittig反应来合成梨小食心虫性外激素,找到了最有利的反应溶剂条件,使产物中的顺反构型产物正好占88%,田间试验表明具有最大的诱蛾活性.这就大大缩短了合成路线,降低了成本,使梨小食心虫性外激素在生产上得到推广使用.3结论综上所述,在有机化学反应中,溶剂不仅影响着反应历程,而且对立体化学也有着重要的影响.因此在有两种和多种可供选择的反应途径中,可通过选择适当溶剂,使反应按某一途径进行,得到高效的产物.参考文献:〔1〕丁新腾,黄乃聚.有机化学[M].高等教育出版社,1985.〔2〕南京大学化学系有机化学教研室主编.有机化学(下册).人民教育出版社,1979. 〔3〕尹冬冬.有机化学(下册)[M].高等教育出版社,2006.〔4〕陈光旭.有机化学专题论(一)[M].北京师范大学出版社,1987.。
高等有机化学课件电子效应和溶剂效应

对于+I效应也是同样规律, Na(0.9)的电负性小于Mg(1.2) ,前者的活性要大得多,同 样,由于Li的电负性(1.0)很小,给电子能力大于Mg,因此有机锂试剂活性大于格氏试剂, 有很强的亲核能力.
(CH3)3C
O
C(CH3)3
+
1) -78oC (CH3)3CLi 2) H+,H2O
[(CH3)3C]3COH
高等有机化学课件电子效应和溶剂 效应
上述有机化合物分子的酸性变化,都是分子中电子云密度分布发生了变化,或者 从另一个角度说键的极性发生变化的结果。有机分子中,由于电负性不同的取代基 的影响沿着键链传递,致使分子中电子云密度按取代基相对于氢的电负性所决定的 方向而偏移的效应,叫做诱导效应。 (I)诱导效应可表示为:
-I: C CR >-CR=CR >-CR2-CR3 =O>-OR N >=NR>-NR2
CH3COCOOH>CH3OCH2COOH
以上讨论的是都是在静态下分子中电子云密度的分布情况的变化,故称为静态 诱导效应。当某一外来试剂的极性中心接近反应分子时,尤其是在试剂分子开始 进攻而导致过渡态的形成时,也会引起分子中电子云的暂时改变,这种改变叫做 动态诱导效应。这里要注意的是在实际反应中,动态的诱导效应往往起着更重要的 作用,并且有助于反应的进行。幸亏,大多数情况下,静态和动态诱导效应的方向 是一致的。
-I 效应:--NO2>-N+(CH3)3>-CN>-F>-Cl>-Br>-I>-OH
+I 效应: -C(CH3)3>CH2CH3>-CH3 注:这是取代乙酸在水溶液中的离解常数确定的,气相中的结果与此有一些不同
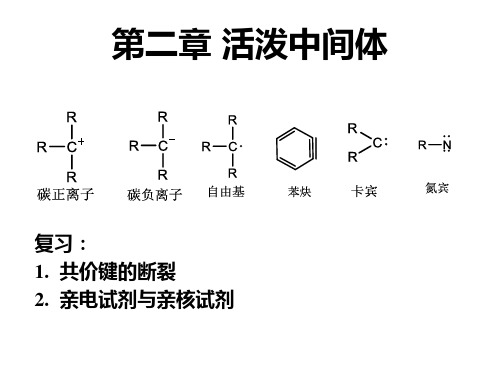
高等有机化学 第二章 有机反应中的活性中间体

.
44
② 碱性条件下脱羧(C—C键异裂)
CN
碱
C 2H 5C COOH
C 6H 5
CN C 2H 5C
C 6H 5
29.04.2020
.
45
(2)负离子对不饱和键的加成
H C C H+O C H 3
C HC HO C H 3 H O C H 3
C H 2 C HO C H 3 + O C H 3
29.04.2020
.
37
5
4 3
SN2
6
1
OBs
exo- 2
54 3
1 6+2
SN1 +
endo-
+
OBs
而内型的几何形状不具备背面进攻的条件,即-
OBs基团阻碍了C-6对C-2的背面进攻,反应按
SN1历程进行,因此速度小,但内型化合物生
成C正离子后能迅速转变成同样的非经典的C正
离29.子04.20,20 因此产物相同。 .
29.04.2020
.
46
(3)来自金属有机化合物 RMgX、RLi、R2CuLi、RZnX
2 R M g XR 2 M g + M g X 2 R 2 M g M g X 2
OEt2 R Mg X
OEt2 R Mg R
OEt2 X Mg X
29.04.2020
OEt2
OEt2
.
OEt2
47
2. 碳负离子的结构 ~109°28′
环庚三烯正离子
环戊二烯正离子
有芳香性,特别稳定 反芳香性,很不稳定
29.04.2020
.
19
空间效应 碳正离子中心碳原子必须sp2杂化,才较稳定。 ①平面构型有利于电荷离域; ②空p轨道的两瓣在平面两侧均可溶剂化。
第二章+空间效应和溶剂效应

第一节 空间效应
一. 空间效应的定义 【定义】:是指分子的空间结构给予分子性质的异常影响。 分子内或分子间不同取代基相互接近时,由于取代 基的体积大小、形状不同,相互接触而引起的物理相互 作用 -空间效应 (位阻效应)。 是一种物理的相互作用。 二. 几种常见的空间效应 1. 空间位阻所引起的各种张力对有机反应活性的影响 ①. 空间位阻引起的B张力效应(back strain 后张力) 【定义】:指空间位阻引起的基态张力反而能增加分子 离解反应速度的效应。
化学与化工学院
在甲醇中,乙酸的酸度(Ka=2.2×10-10)约为吡啶翁离 子(Ka=2.8×10-6)的万分之一。 (2). 溶剂化引起互变异构平衡反应平衡常数的改变 例如:乙酰乙酸乙酯的酮式和烯醇式的互变平衡中:
H3C C O CH2 OEt C O K 25℃ H3C C O H H C C O OEt
主
C H3 H 3C H3C H O Li Al H 4 t-Bu O H H3C H3C C H3 H H OH H3C H3C
次
C H3 H OH H
次
化学与化工学院
主
6. 空间位阻引起单键正常旋转受阻 空间位阻往往会阻碍单键正常旋转受阻而导致两种情 况的发生。 一是:分子对称性消失,产生了旋光异构; 二是:使分子具有类刚性结构,产生了顺反异构体。
+
HC l
化学与化工学院
总结论:空间效应对化合物的性质,尤其在反应过程中形成 的活性性中间体的稳定性影响是较大的,对有机反应的进 程起着重要的作用。
化学与化工学院
第二节
一、溶剂化的定义及其特点
溶剂效应
【定义】:由于溶剂化而给予反应物分子性质的异常影响及反 应现象异常变化的现象叫做溶剂效应。
第二章 有机反应中的活性中间体详解
(3)溶剂化效应 溶剂化效应对碳正离子的稳定性影响比较严重,含有孤对电 子的极性质子溶剂能够很好的将碳正离子溶剂化,从而稳定 性增强。
常见的极性非质子溶剂: 四氢呋喃(THF),二甲亚砜(DMSO),丙酮,乙腈,二甲基甲酰胺 (DMF),二甲基乙酰胺(DMAC). 常见的质子性溶剂: 常与溶质分子以氢键缔合的溶剂,一般含有羟基、氨基或羧 基的化合物。如:水、乙醇、甲酸、乙酸、乙胺。
3.重排:烷基、芳基、氢或其他基团,带着它的一 对成键电子迁移到正离子中心碳原子上,使迁移的 起点碳原子上带正电荷。迁移的总结果是由比较不 稳定的碳正离子产生比较稳定的碳正离子。 举例:
Pinacol重排
邻二醇在酸催化下,经过碳正离子的重排成为酮
或醛的反应。
Wagner-Meerwein重排
醇羟基的β位上是个仲碳原子 或叔碳原子时,在酸催 化脱水中,常常会发生的重排反应。
pKDMSO
17.2
24.7
三、碳负离子的形成
• 1.R-H解离: • 一般在强碱作用下便可生成碳负离子。
2. 亲核加成反应
3. 生成金属炔化物或带负电荷的芳香化合物
4. 格式试剂(极性转化)
四、 碳负离子的反应类型
1.亲核取代反应:烷基化和酰基化
举例:
2. 缩合反应
克莱森(Claisen)酯缩合反应—乙酰乙酸乙酯的合成
CF3CH3
CH3CH3
(4) 共轭效应
>
O
O
O
O
(5) 芳香性
2-
2-
(6)溶剂化作用 碳负离子的溶剂化作用主要是形成氢键的影响,因此极性 非质子溶剂中更加活泼。而在质子性溶剂中往往形成氢键 而比较稳定。 C-H酸 O2NCH2NO2 CH3COCH2COCH3 CH3NO2 PhCOCH3 pKH2O 3.6 9 10.2 15.8
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
例:CaCl
在水-甲醇体系中,Ca2+和Cl–都优先被
2
水溶剂化。
如阳离子优先被一种溶剂溶剂化,而阴离子优先被另一种溶剂溶剂化,则称异选择性溶剂化。
例:硝酸银在乙腈-水体系中,Ag+优先被乙腈溶剂-优先被水溶剂化。
化, 而NO
3
2、溶剂和溶质分子间的相互作用
第一类包括定向诱导力和色散力,这些力是非特异性的,不可能完全饱和。
第二类包括氢键力和电荷转移力,或称电子对授受力。
这类作用有方向并且可以饱和生成化学计量的分子化合物。
C、偶极-诱导偶极力
具有永久偶极矩的分子或离子能诱导邻近分子,产生诱导偶极矩,分子在被诱导的瞬间总是处于诱导偶极的方向,两者之间有吸引力。
非极性分子可极化率越大,诱导偶极矩也越大。
这对偶极分子和离子在非极性溶剂中的体系最重要。
D、瞬间偶极-诱导偶极力(色散力〕
非极性分子由于电子不断运动,会瞬间产生小的偶极矩,它使邻近分子产生脉冲性极化,从而产生分子间的相互吸引力,这称为色散力。
在两个键已饱和的分子间形成一个附加的成键作用必须是在电子给体分子中存在一个能量足够高的已占据分子轨道,而在电子受体分子中存在一个能量足够底的未占据分子轨道。
3、溶剂的极性和分类
(1)质子性溶剂
(2)极性非质子性溶剂
(3)非极性溶剂
量度溶剂极性的标准:
(1)偶极矩u
常见有机溶剂分子偶极矩的数值在0-5.5D, 在不存在特异性溶质-溶剂间相互作用时,分子极性大小与偶极矩大小一致。
溶剂极性加大,K T 值降低,cis-烯醇式减少。
因为:1、在两种互变异构体中,烯醇式极性较小。
分子内氢键的形成降低羰基偶极-偶极斥力,而在酮式中,这种斥力使其极性加大。
2、分子内氢键使烯醇稳定化,溶剂极性加大,分子间氢键加强,分子内氢键被削弱,烯醇含量减少。
烯醇含量与1,3-二羰基化合物浓度有关。
当偶极性的1,3-二羰基化合物用非极性溶剂稀释,溶剂与羰基作用弱,两羰基偶极斥力大,不稳定;烯醇与溶剂分子间氢键弱,分子内氢键强,烯醇含量增高。
用偶极溶剂稀释,烯醇含量降低。
溶剂影响反应速率的定性理论-休斯-英戈尔德(Hughes-Ingold)规则:
1、如活化络合物比反应物具有更大电荷密度,则提高溶剂极性将使反应速度加快。
2、活化络合物比反应物电荷密度小,则提高溶剂极性将使反应速度降低。
3、如从反应物到活化络合物的转变电荷密度变化很小或没有变化,则溶剂改变对反应速率影响很小。