高斯软件的介绍共43页
高斯的介绍和使用

AO轨 道
轨道系数 LUMO
注: • 输出的MO系数是未经归一化过的; • 1s, 2s等符号并非真正意义上的AO, 它与计算 所采用的基组 有关. 对于上例, 采用的是6-31G基组, 可知, 对于价层AO是 分裂为两组的, 故对于O原子, 输出的2s和3s 实际上均为价 轨道, 其它类推. • 当基函数较多时, 若只考察前线附近的轨道成 分, 可用关键 词: pop=regular, 此时只给出5个占据轨道和5 个空轨道组成.
距离矩阵
各原子的直角坐标
需核查!
注意:计算结果是以该坐标系为准
需核查!
MO初始猜测
迭代次数 收敛指标
自洽场迭代求解部分
迭代次数 收敛指标
自洽场迭代求解部分
偶极矩及多极矩
对主要计算结果进行总结
分子轨道系数的输出:关键词: pop=full说明: 有时为 了分析MO成分, 则需利用该关键词输出各MO的成 分, 以H2O为例:
Mulliken电荷分布
说明: 电荷的绝对值是没有意义的,其数值受到所用方法, 尤其是 基组的影响较大: 以H2O为例: 方法/基组 B3LYP/STO-3G B3LYP/3-21G B3LYP/6-31G HF/6-31G O H
-0.329 -0.637 -0.705
-0.795
0.165 0.318 0.353
二、安装高斯对计算机的要求
现在的 PC 速度非常快,成本低廉,是使用 Gaussian 的一 个经济实惠的平台。 对稍懂计算机的人我会建议组装一台 Linux 工作站,基本的配备应包括 64 位 Dual-Core CPU, 如 Intel P4 8xxD, 9xxD, Core Duo, 或 AMD Athlon 64X2 等, 2 GB DDR2 667 以上内存, 2 台 200 GB 以上 SATA 硬盘以 弹 性 使 用 Raid 0/1 。 Linux 操 作 系 统 可 选 择 免 费 下 载 之 Fedora Core 4 或 5,Suse 10 等,记得要使用 64 位的版本。 旧的 32 位 PC 也可以用,只是效能上会差很多。接下来要依 照你的系统种类选择适当的 Gaussian 03 版本,请参阅本站 首页。目前 Window XP 的版本 (G03W) 还不能发挥 64 位的 运算效能, 使用上的便利性及功能性也都不如 Linux 的版本
高斯软件-基础教程
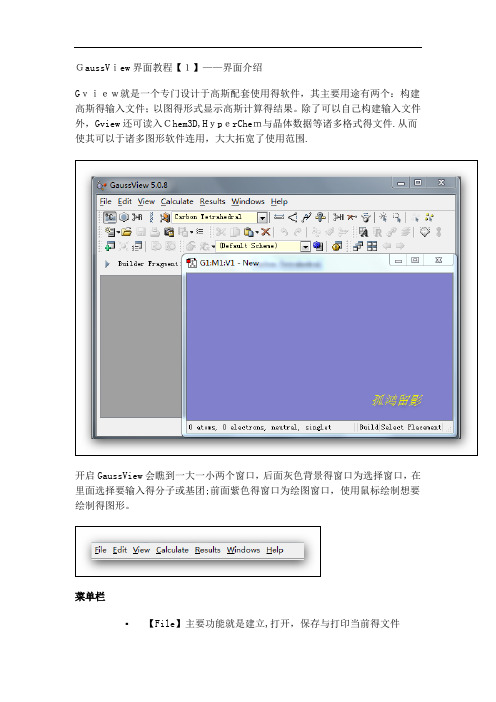
GaussView界面教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件.从而使其可以于诸多图形软件连用,大大拓宽了使用范围.开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
《高斯软件入门》课件

统计分析实例
通过实例深入理解高斯软件 在统计分析方面的应用和技 巧。
总结与展望
高斯软件的优越性
总结高斯软件的优势和特点,以及为什么它是数据分析的理想选择。
高斯软件的发展趋势
展望高斯软件未来的发展方向和趋势,激发你对技术的好奇心。
学习高斯软件的建议与方法
提供一些建议和学习方法,帮助你更高效地学习和掌握高斯软件。
高斯软件的优点和局 限性
讨论高斯软件的优点和局 限性,帮助你更好地了解 该软件的适用范围和限制。
高斯软件安装与环境配置
1 安装高斯软件的步骤
详细介绍高斯软件的安装过程,帮助你顺利完成安装。
2 配置系统环境变量
指导你如何配置系统环境变量,确保高斯软件可以在你的计算机上正常运行。
3 检查安装是否成功
提供一些简单的检查方法,确保你成功安装了高斯软件。
高斯软件基础知识
1
高斯软件的数据类型
介绍高斯软件支持的不同数据类型,如数值、字符和逻辑。
2
高斯软件的数组与向量
讲解高斯软件中数组和向量的定义、操作和应用。
3
高斯软件的函数与运算符
探索高斯软件提供的丰富函数和运算符,以及它们在数据处理中的作用。
高斯软件高级应用
高斯软件的图形绘制功能
展示高斯软件强大的图形绘制 功能,帮助你更直观地呈现数 据。
高斯软件的数据处理功能
介绍高斯软件用于数据
介绍高斯软件在统计分析方面 的功能和应用,助你深入理解 数据潜在的规律。
高斯软件编程实例
矩阵运算实例
通过实例演示高斯软件中矩 阵运算的基本操作和应用。
数据拟合实例
展示如何使用高斯软件进行 数据拟合和曲线拟合,帮助 你更好地分析数据模型。
高斯软件-基础教程

GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【File】主要功能是建立,打开,保存和打印当前的文件▪【Edit】完成对分子的剪贴、拷贝、删除、抓图等▪【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后的结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
高斯软件-基础教程

GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
GAGGAGAGGAFFFFAFAF开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
GAGGAGAGGAFFFFAFAF菜单栏【File】主要功能是建立,打开,保存和打印当前的文件【Edit】完成对分子的剪贴、拷贝、删除、抓图等【View 】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等【Calculate】直接向Gaussian提交计算【Results 】接收并显示Gaussian计算后的结果【Windows】控制窗体,如关闭、恢复等【Help】帮助快速工具栏GAGGAGAGGAFFFFAFAF【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
GAGGAGAGGAFFFFAFAF【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;GAGGAGAGGAFFFFAFAF【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等GAGGAGAGGAFFFFAFAF【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】GAGGAGAGGAFFFFAFAF这里面的所有选项都可以通过在绘图窗口点击右键得到。
高斯软件的介绍

题 目:高斯软件的简介 指导老师:徐畅老师 姓 名:马乃宇 学 号:C13201045 专 业:高分子化学与物理专业
Gaussian是一个量子化学软件包,它是目前 应用最广泛的计算化学软件之一,Gaussian 软件的出现降低了量子化学计算的门槛, 使得从头计算方法可以广泛使用,从而极 大地推动了其在方法学上的进展。
4).最简单的关键词输入是#或#p,其含义是采用HF方法和 STO-3G基组计算体系的能量;
能量的计算: 如何计算一个体系的能量是获取分子各种性质的基础,因此 首先来看如何计算体系的能量,即进行单点能计算: (1). 计算方法的选择: g98提供的常用计算方法有: 1) 半经验方法: 关键词:AM1, PM3, CNDO, INDO, MINDO 它们主要用于大的有机分子体系(由上百个原子组成),一般 对于含金属体系不适用。这些方法只有在特殊场合适用。 2) 从头算(ab initio)方法: HF方法:即基于Hartree-Fock原理的方法 关键词:HF,RHF,UHF,ROHF 说明:I)当关键词为HF时,会自动根据自旋多重度选择 RHF还是UHF; Ii)ROHF为限制性开壳层HF方法,与UHF区别在 此时除了成单电子外,其余的和电子仍配对, 通常该方法得到的能量要较UHF略高。 Iii)HF方法可以看作是最低级的从头算方法,该方 法除了在构型优化时有使用外,不适合计算能量。
(2).基组的选择: 1). 全电子基组: 关键词:sto-3g, 3-21g, 4-31g, 6-21g, 6-31g, 6-311g, d95/d95v 说明:I). 不同的基组适用范围是不同的: STO-3G(H-Xe);3-21G(H-Xe);6-21G(H-Cl) 4-31G(H-Ne);6-31G(H-Kr);6-311G(H-Kr) D95(H-Cl 除了Na, Mg);D95V(H-Ne)
高斯软件基础教程
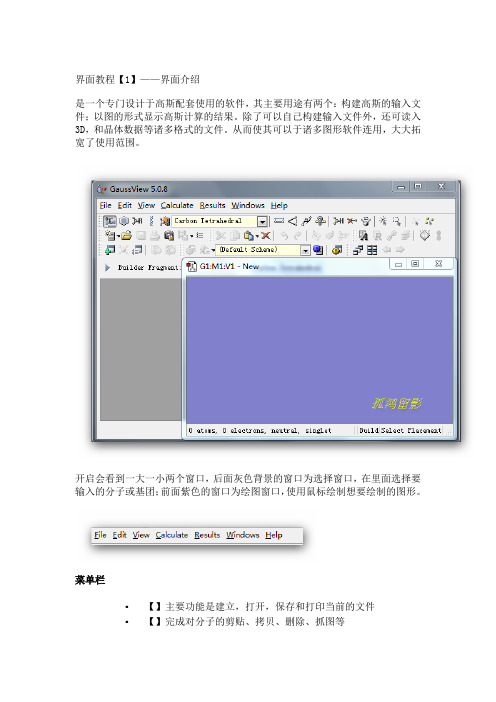
界面教程【1】——界面介绍是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,还可读入3D,和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏▪【】主要功能是建立,打开,保存和打印当前的文件▪【】完成对分子的剪贴、拷贝、删除、抓图等▪【】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【】直接向提交计算▪【】接收并显示计算后的结果▪【】控制窗体,如关闭、恢复等▪【】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从里递交计算为例来说明。
1、开启2、双击按钮,选择苯基(第一个),会看到主程序框体中出现苯环在工作窗口单击,可以看到工作窗口也出现了一个苯环。
单击左键,可以将主程序框体中的分子或基团加入到工作窗口;按上下左右键,或者左键单击不放移动鼠标,可以调节分子的角度;滚动鼠标滚轮,可以放大缩小分子;按住键,左键单击不放移动鼠标,可以移动分子;当工作窗口内有多个分子时(在构建大的分子时,这种情况很容易出现)用鼠标左键组合,移动想要移动的分子;用鼠标左键组合调节其中一个分子的角度;双击图标(或者左键不放),在元素周期表中选择【F】元素,回到工作窗口在苯的任意一个【H】上单击,使之变成【F】双击图标,从链烃库中选择【乙基】(第一个),然后点击间位上的【H】即可至此,分子式已经构建完成,存储个人常用分子的功能,双击上的图标,在下面的对话框中键入相关项目,保存即可。
2.高斯软件简介

每个文件一个 分子集合 所有文件做成 一个分子集合 用所有文件创 建一个分子
GaussView简介
File菜单:文件打开保存
【New】 新建分子模型,有两种创建形式: (1)创建新的分子集合; (2)在当前分子集合中添加新的分子。
打开已有模型和其他输出文件 快速打开最近打开过的文件; 快速打开与当前活动分子相关的文件
• 冗余坐标编辑器
用于创建和编辑冗余坐标,冗余坐标常用于Gaussian 优化 或势能面扫描中。
• 分子轨道编辑器
注意需从检查点文件中读入文件。
GaussView简介
计算工具栏【Calculate】
◆创建 Gaussian 作业
【Calculate】→【Gaussian Calculation Setup】
GaussView简介
键盘与鼠标配合
当窗口中有多个分子, 只想转动或者移动其中一 个分子, 按住键盘【Alt】键
1. 2. 用鼠标左键点击某个分子并按住,此时移动鼠标,只有 被点击的分子会转动; 用鼠标中键(或滚轮)点击某个分子并按住,移动鼠标, 只有被点击的分子会跟随鼠标移动
3.
4.
用鼠标右键分子点击某个分子并按住,左右移动鼠标, 只有被点击的分子会沿一个垂直屏幕的轴转动;
2.
3.
GaussView简介
练习2
• 通过转动二面角将全重叠式丁烷转成全交叉式 全重叠式 全交叉式
GV_exercise_2.gjf
GV_exercise_2_OK.gjf
GaussView简介
Builder面板一些图标的意义
查询
选择所有的原子 不选择任何原子
重新键和
添加一个键合原 子(默认添加H) 删除一个原子