GaussView使用简介
高斯软件-基础教程
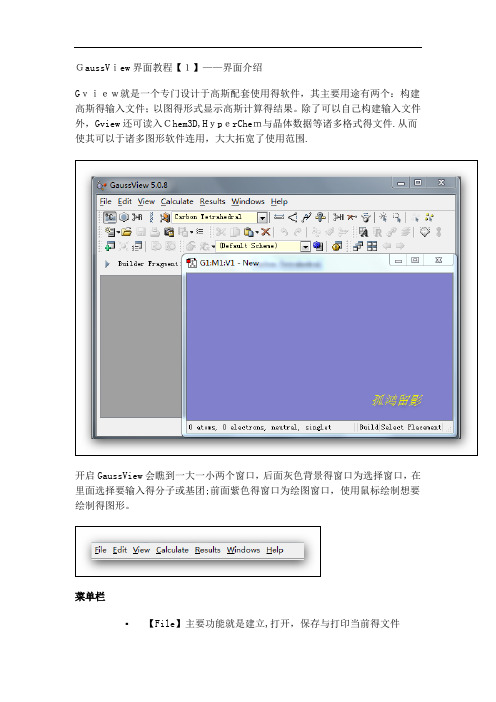
GaussView界面教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件.从而使其可以于诸多图形软件连用,大大拓宽了使用范围.开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
GaussView基础教程
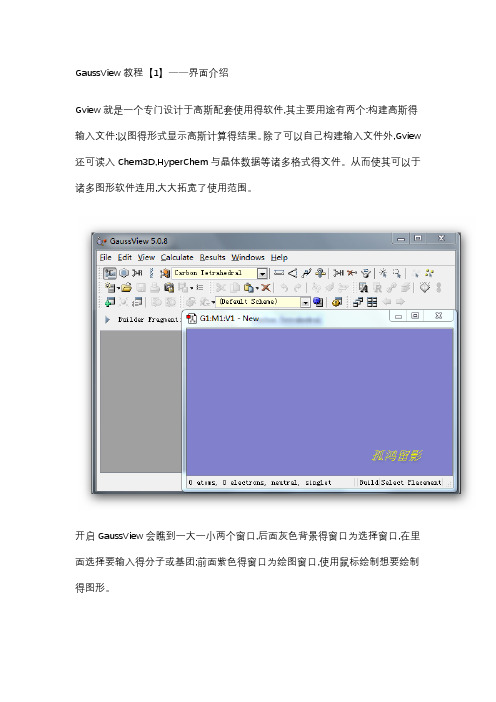
GaussView教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview 还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
GaussView基础教程
GaussView教程【1】——界面介绍Gview就是一个专门设计于高斯配套使用得软件,其主要用途有两个:构建高斯得输入文件;以图得形式显示高斯计算得结果。
除了可以自己构建输入文件外,Gview 还可读入Chem3D,HyperChem与晶体数据等诸多格式得文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会瞧到一大一小两个窗口,后面灰色背景得窗口为选择窗口,在里面选择要输入得分子或基团;前面紫色得窗口为绘图窗口,使用鼠标绘制想要绘制得图形。
菜单栏▪【File】主要功能就是建立,打开,保存与打印当前得文件▪【Edit】完成对分子得剪贴、拷贝、删除、抓图等▪【View】与显示分子相关得都在这个菜单下,如显示氢原子、键、元素符号、坐标等▪【Calculate】直接向Gaussian提交计算▪【Results】接收并显示Gaussian计算后得结果▪【Windows】控制窗体,如关闭、恢复等▪【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会瞧到一个元素周期表,通过它可以选择需要绘制得元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只就是这里提供得都就是环状化合物残基;【左面第三个】提供常用得R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用得基团存放到此处这条快速编辑栏中从左到右依次就是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面得所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏就是最常用得,几乎所有软件都有得新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
Gaussview软件使用手册

Gaussview软件使用手册GaussView就是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView就是不在高斯软件的计算模块中,而就是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至就是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译与放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其她的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1.经过优化的分子结构;2.分子轨道3.任何估算密度的电子密度表面;4.静电潜在表面;5.原子价;6.与振动频率相符的一般模型;7.IR,Raman,NMR,VCD以及其她的频谱;8.结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP与BOMD轨道的模拟;9.整体能量的小部分以及其她来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目就是按照逻辑结构来排列的,开始于最简单的项目,然后就是引用最广泛的项目,最后就是更加复杂与不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者就是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。
Gaussview软件使用手册

Gaussview软件使用手册GaussView是一个被设计来帮助准备输入进入高斯软件的文档以及以图形的形式来检验高斯软件完成并输出的结果的图形化的用户界面。
GaussView是不在高斯软件的计算模块中,而是以前端/后端的模式来协助高斯软件的运行。
GaussView为高斯用户提供了三种主要的便利。
首先,通过它先进的可视化工具,GaussView使得用户可以很快的做出大分子的结构图甚至是很大的分子,然后旋转,在这些分子的基础上经过简单的搜寻操作来编译和放大。
它同样可以载入例如PDB类型的标准的分子结构文档。
第二,GaussView使得建立多种形式的高斯计算变得容易。
它使得准备为普通工作模式以及如ONIOM,QST2/OST3转变结构优化方法,CASSCF计算方法,周期边缘状态计算方法(PBC)以及其他的许多前沿方法的复杂的载入更加简单。
如果高斯软件被安装在了同一台电脑上的话,用户同样可以使用GaussView来进行工作。
最后,GaussView使得用户可以通过多种的图形技巧来检验高斯计算的结果。
高斯的计算结果中可以用图形来表示的有下列几种:1. 经过优化的分子结构;2. 分子轨道3. 任何估算密度的电子密度表面;4. 静电潜在表面;5. 原子价;6. 与振动频率相符的一般模型;7. IR,Raman,NMR,VCD以及其他的频谱;8. 结构优化,IRC反应途径的后续,能级表面的扫描以及ADMP和BOMD轨道的模拟;9. 整体能量的小部分以及其他来自于相同工作类型的数据;这本书为所有的GaussView结构提供了一个参考。
该软件的每一个程序的结构都被详细记录。
将项目按照一般目的分类。
在每一个分类中,项目是按照逻辑结构来排列的,开始于最简单的项目,然后是引用最广泛的项目,最后是更加复杂和不经常使用的项目。
为了找出用户感兴趣的特殊信息,可以通过索引来打开有关主题或者是特殊的对话框。
颜色收集以及对话在索引中的主题菜单下(下面直接中英语)显示了出来。
gaussview使用简介

(12).向Gauss递交计算。点Gview界面上Calculation 会出来一种递交计算旳对话框。从所给旳对话框中能够选择工作类型Job Type(如优 化,能量或频率等);计算措施Method(如半经验措施,HF措施,DFT措施,MP措施 等,还能够选定基组);Title(对所要做旳计算给一种阐明,以备后来旳查看) Link 0(给 检验点文件命名,还能够在此用RWF命令设置临时数据互换文件旳大小); General, Guess,(这两个选项主要是给出体系中各原子旳连接关系及怎样给出初始猜测); NBO(可在此设定NBO计算),PBC(可在此设定晶体旳有关计算), Solvation(可在 此设定溶液中旳计算,除了选择溶剂外,还要选择模拟溶剂旳理论模型)旳Gaussian
打开Gview ,下图就是Gview打开后旳窗口
(2).双击窗口中 图标,得到如下窗口里面有常用旳环状官能团。选中苯环(单 击即可选中)
(3).在目前工作窗口(打开Gview时程序自动打开一种工作窗口,如下图)也可经过 File-new 途径 新建一种工作窗口
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图
GaussView使用简介
Gview是一种专门设计于高斯配套使用旳软件,其 主要用途有两个构建高斯旳输入文件以图旳形式显示 高斯计算旳成果除了能够自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式旳文件。从而使其能够于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
在构建分子前,应对分子旳键长,键角和点群等有一种详细旳了解。这么在构建过 程中才干更加好旳把握细节,如构建大分子时,各个集团间旳键长,键角和二面角 一定要给旳精确。相当一部分旳错误(如SCF不收敛,1502报错等)都是分子建模 不合理造成旳。分子模型不合理造成程序根据分子模型所得旳薛定鄂方程不合理, 也就造成了对方程旳解不能顺利完毕,所以报错。对于大分子,不会出现分子直线 排步旳情况,链烃一般为锯齿型旳排列,环之间总是有一定旳夹角,共面情况虽有 但不多见,这些都是在构建分子模型旳过程中需要注意旳。再者,分子建模是量化 计算旳一种难点,尤其是对金属配合物,块状体系和纳米体系旳构建。只有给出合 理旳构造,程序才干给出合理旳解。假如不能体现出分子旳对称性,则程序不能正 确判断分子轨道旳对称性,这在涉计到反应机理和轨道相互作用旳研究中可能会造 成错误旳判断。
gaussview使用教程1

• 元素板块
• 在周期表中选择元素种类,以及原子的成键 方式
• 端基板块提供了一些列已经过AM1方法优化的端 基碎片,默认端基为羰基。
• 环形板块提 供了一系列 AM1方方优 化后的环形
结构,默认 环为苯。
• 生物分子下拉菜单可选择所需的是氨基酸或核苷。
• 当选择氨基酸时,左边下拉菜单可选择氨基酸种 类,右边下拉菜单选择分子片断作为中间部分、 氨基端或酰基端。这样就制定了氨基酸的种类。
• Always track point group symmetry:当结构变化时,不断计 算分子所属点群,此选项与对称性限制为None联合使用,当 改变键长的结构参数时,可以看到对称性的变化,通常来说, 只有参数的改变完成后,对称性才会发生改变,对一些大的 体系,此选项可能导致反应时间明显变长。
Bond SmartSlide对话框也可以在没有化学键相连的原子间添 加化学键。当对话框打开时,可通过单击选择所需要的化学 键类型。同样也可以取消原子间的化学键。
• Displacement选项指认当键长改变时,原子所连接的基团的 变化。
• Translate Atom:只移动原子,所连接基团的位置不变。 • Translate Group:原子所连接基团的位置随原子改变(即看
• Rebonding Structures • Rebonding按钮和Edit-Rebond选项:以间距算法为
基础确认原子间距离,Gaussian不用屏幕所显示的 键长信息来计算,这些信息是为了便于我们观看。
Imposing Symmetry对话框
• Imposing Symmetry
• Enable point group symmetry:使Gaussview图形具有对称 性。
高斯软件基础教程

高斯软件基础教程 Revised by BLUE on the afternoon of December 12,2020.GaussView界面教程【1】——界面介绍Gview是一个专门设计于高斯配套使用的软件,其主要用途有两个:构建高斯的输入文件;以图的形式显示高斯计算的结果。
除了可以自己构建输入文件外,Gview还可读入Chem3D,HyperChem和晶体数据等诸多格式的文件。
从而使其可以于诸多图形软件连用,大大拓宽了使用范围。
开启GaussView会看到一大一小两个窗口,后面灰色背景的窗口为选择窗口,在里面选择要输入的分子或基团;前面紫色的窗口为绘图窗口,使用鼠标绘制想要绘制的图形。
菜单栏【File】主要功能是建立,打开,保存和打印当前的文件【Edit】完成对分子的剪贴、拷贝、删除、抓图等【View】与显示分子相关的都在这个菜单下,如显示氢原子、键、元素符号、坐标等【Calculate】直接向Gaussian提交计算【Results】接收并显示Gaussian计算后的结果【Windows】控制窗体,如关闭、恢复等【Help】帮助快速工具栏【左面第一个】选择元素与价键,单击打开会看到一个元素周期表,通过它可以选择需要绘制的元素以及价态。
【左面第二个】环工具,作用与上一个差不多,只是这里提供的都是环状化合物残基;【左面第三个】提供常用的R基团模板,其中包括乙基、丙基、异丙基、异丁基等【左面第四个】氨基酸残基,使用它可以迅速绘制氨基酸【左面第五个】用户自定义基团,您可以将常用的基团存放到此处这条快速编辑栏中从左到右依次是【键调整】|【键角调整】|【二面角调整】|【查询已有结构】|【增加化学键】|【删除化学键】|【翻转原子】|【单个选择】|【框选】|【去除选择】|【全选】这里面的所有选项都可以通过在绘图窗口点击右键得到。
3、常用工具栏这两条条工具栏是最常用的,几乎所有软件都有的新建打开等工具GaussView教程【2】——构建分子这里以构建一个间氟苯乙烷分子并从GaussView里递交计算为例来说明。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
(4).双击Gview界面上的 图标。出现以下窗口点击氟的元素符号“F”,就选中氟原子。
(5).回到工作窗口在苯环上的任一个H上单击左键,将H置换成F。
但要注意此时仅是元素符号发生了改变,C-F间的距离仍是C-H键的距离。需要用建 长工具进行调整。单击Gview界面上 图标。 然后再点击工作窗口中的C原子和F原 子,看到被选中的两个原子于周围的原子再亮度上有差异此时会出现下面的窗口
(12).向Gauss递交计算。点Gview界面上Calculation 会出来一个递交计算的对话框。从所给的对话框中可以选择工作类型Job Type(如优化, 能量或频率等);计算方法Method(如半经验方法,HF方法,DFT方法,MP方法等, 还可以选定基组);Title(对所要做的计算给一个说明,以备以后的查看) Link 0(给检查 点文件命名,还可以在此用RWF命令设置临时数据交换文件的大小); General, Guess,(这两个选项主要是给出体系中各原子的连接关系及如何给出初始猜测); NBO(可在此设定NBO计算),PBC(可在此设定晶体的有关计算), Solvation(可在 此设定溶液中的计算,除了选择溶剂外,还要选择模拟溶剂的理论模型)的Gaussian
(10).查看分子的对称性 从Edit-Point group路径可以查看所构建分子的点群。点击Point group后,出现如下 窗口:为C1点群,其下拉菜单中的为可能的点群(改变Tolerance,也可帮助我们判断 所构建体系可能有的点群)
(11).查看分子坐标。单击Gview界面上的 图标。出现下面窗口 Z表示时内坐标,C表示直角坐标。可以在里面对坐标做适当的调整
(8).单击Gview界面上 图标。然后点击与乙烷正对的苯上的C原子和H原子选择 “None”,乙烷上的C-H键作类似处理然后单击Gview上的 图标,点选工作窗口中的 两个氢原子,即将它们删去
至此分子构建已完成。
9).Gview存储个人常用分子的功能。Gview中只有常用的一些环状分子和链状分子, 远不能满足研究特定体系的特殊需要。不过Gview有一个功能可以弥补这个缺憾:可 以把常用的分子或官能团存在制定的文件夹内。在需要时可以直接调用。双击view上 的 图标,在下面的对话框中键入相关项目,保存即可
Gview的界面及主要功能键的介绍
GVIEW的界面
第一行为菜单栏:
File:主要功能是建立,打开,保存和打印当前的文件 Save Image 将当前文件保存为图片格式 Preferences。可以在里面改变Gview默认的各种显示的设置。
Edit: 在这里可以完成对分子的剪贴,拷贝,删除 和 抓图等。 Atom List,显示当前分子的内坐标,笛卡儿坐标,分数坐标等。 Point Group 可以显示当前分子的点群及可能有的点群。 PBC显示晶体文件(可以将CIF文件转换为图形,在点PBC按钮后所给并的对话框 中根据选项调节具体显示的格式。 Mos用于显示分子轨道(只有检查点文件,此选项才能给出分子轨道图。 Symmetrize,对当前体系进行对称性控制。
Result :显示计算的结果,包括电荷,静电势表示的表面,振动,频率,核磁,势能面 扫描,优化等。注意有些结果只能 用检查点文件才能显示。
常用功能键的介绍
以构建一个间氟苯乙烷分子并从 Gv Nhomakorabeaew里递交计算为例来说明
(1)双击Gview图标, 打开Gview ,下图就是Gview打开后的窗口
View 这里面的选项都是于分子的显示有关的,如显示氢原子,显示键,显示 元素符号,显示坐标轴等
可从Gview中直接向高斯提交计算。这是Gview作为高斯软件配套功能的重要体现。 从所给的对话框中可以选择工作类型Job Type(如优化,能量或频率等);计算方法 Method(如半经验方法,HF方法,DFT方法,MP方法等,还可以选定组);Title(对所 要做的计算给一个说明,以备以后的查看) Link 0(给检查点文件命名,还可以在此用 RWF命令设置临时数据交换文件的大小); General,Guess,(这两个选项主要是给出体 系中各原子的连接关系及如何给出初始猜测);NBO(可在此设定NBO计算),PBC (可在此设定晶体的有关计算), Solvation(可在此设定溶液中的计算,除了选择溶剂 外,还要选择模拟溶剂的理论模型)
根据C-F键的长度在 0.675和2.700之间的方框内进行C-F键的调整。完毕后点击OK即可。
(6).双击Gview界面上的 图标,出现以下窗口这是Gview里内置的链烃库,选中 乙烷
(7).在工作窗口内空处点左键,用我们前面讲过的命令调节苯和乙烷间的距离和角度
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度;将鼠标放在分 子上,前后移动,可以将分子放大或缩小。Shift+Alt+鼠标左键组合可以在窗口内平 移分子当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可 用以下命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间 的距离。可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子 间的角度。Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移 动,可以调节分子的角度]这个功能连用。具体的好处需要自己去试
GaussView使用简介
Gview是一个专门设计于高斯配套使用的软件,其 主要用途有两个构建高斯的输入文件以图的形式显示 高斯计算的结果除了可以自己构建输入文件外, Gview还可读入CHEM3D,HYPERCHEM和晶体数据 等诸多格式的文件。从而使其可以于诸多图形软件连 用,大大拓宽了使用范围(详见下图)
将鼠标放在分子上,按左键左右或前后移动,可以调节分子的角度将鼠标放在分子上, 前后移动,可以将分子放大或缩小Shift+Alt+鼠标左键组合可以在窗口内平移分子 当工作窗口内有多个分子时[在构建大的分子时,这种情况很容易出现]这时可用以下 命令可以用Shift+Alt+鼠标左键组合移动想要移动的分子,以调节各个分子间的距离 可以用Ctrl+Alt+鼠标左键组合调节其中一个分子的角度,以调节各个分子间的角度。 Ctrl+Alt+鼠标左键这个组合常和[将鼠标左键放在分子上,左右或前后移动,可以调 节分子的角度]这个功能连用。
打开Gview ,下图就是Gview打开后的窗口
(2).双击窗口中 图标,得到如下窗口里面有常用的环状官能团。选中苯环(单 击即可选中)
(3).在当前工作窗口(打开Gview时程序自动打开一个工作窗口,如下图)也可通过 File-new 路径 新建一个工作窗口
在这个窗口中点鼠标左键窗口中就会出现苯分子,见下图