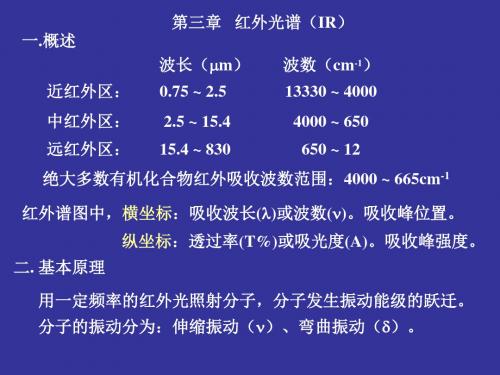
第三章 红外吸收光谱
红外光谱(IR)分析

4. 空间效应: (1)环状化合物的环张力效应:环张力越大,羰 基C=O频率越高。 环张力 四元环 五元环 六元环 (2)空间位阻效应:空间位阻使羰基与双键之间 的共轭受限制,故使C=O频率增高。 5. 氢键效应:氢键的形成,通常可使伸缩振动 频 率向低波数方向移动。
6. 振动偶合效应:当两个基团靠得很近时,产 生振动相互作用,使吸收峰发生分裂。
第三章 红 外 吸 收 光 谱 法
Infrared Absorption Spectrometry
§1 关于红外光谱
红外光谱在可见光区域微波区之间,其波长范 围约为0.75~1000m。
分为三个区: ◆近红外区 0.75~2.5m; ◆中红外区 2.5~25 m; ◆远红外区 25~1000 m
若分子由N个原子组成,则 需3N个坐标(自由度)确定N个原子位置; 分子自由度总数=平动、振动、转动自由度 总和 故 3N=平动自由度+转动自由度+振动自由度 即 振动自由度=3N-(平度自由度+转动自由度) 问题:怎样确定一个分子的平动自由度和 转动自由度?
(1) 平动自由度:分子的质心可沿x、y、z三 个坐标轴方向移动,故平动自由度=3。
2. 共轭效应(C效应):该效应使共轭体系具有 共平面性,电子云密度平均化,造成双键略有 伸长,单键略有缩短。故双键的吸收峰频率向 低波数方向移动。
例. C=O C=O 1715 cm-1 1685~1665 cm-1
3. 中介效应(M效应): 例. C=O 在1680cm-1附近。 若用诱导效应看,则电负性大的N原子应使 C=O键力常数增加,吸收峰位应大于1715cm-1; 但实际情况相反,这是因中介效应造成的。 即N原子上的孤对电子与C=O的电子发生重 叠(p- 共轭),使电子云密度平均化,造成C=O 键力常数降低,故使吸收峰频率移向低波数。
波谱分析 第三章 03红外谱图解析

C= C : 16பைடு நூலகம்0~1450 cm-1区域吸收峰的强弱及个数与分子结 构有关,是判断苯环存在的主要依据。 有2-4个峰,峰数取决于取代基对苯环对称性破坏的程度
苯 甲苯 取代基与苯环共轭时 烷基存在时 1600 cm-1 无吸收 1500 、 1600 cm-1 有吸收 1580 cm-1 处出现强吸收 1450 cm-1 有吸收
烷烃
表3-4, (CH2)n结构 中亚甲基面内摇 摆振动
CH3 –CH – CH2 –CH – CH3 CH3
1168 1386 1468 1367
CH3
2,4 - 二甲基戊烷
78
2.烯烃
基团 = CH中C-H C=C骨架
3080 2975 1680~1620
1000~800 (面外摇摆)
(1) = CH >3000 cm-1为不饱和碳上质子振动吸收,是与饱 和碳上质子的重要区别。 (2) C=C的 位置及强度 与烯碳的取代情况及分子对称性 密切相关。 末端烯烃 C=C吸收最强,双键移向碳链中心时结构对称 性增强, C=C带减弱。顺式较反式强。 共轭双键中由于双键的相互作用出现两个 C=C (1650、1600 cm-1 )。 46
饱和环醚: 在 1260~780 cm-1 范围出现两条或两条以上的吸收带。 环张力增加as波数降低, s波数升高。 O 1071 913 O 983 1028
(3) CH(面外)最有用。 特点是: 不同类型的烯烃,有其独特的波数,且比较固定,不受 取代基的变化而发生很大的变化。 吸收强度特别强。 根据烯氢被取代的个数、取代位置及顺反异构的不同, 出峰的个数、位置及强度不同。
烯烃类型 R1CH=CH2 R1R2CH=CH2 R1CH=CHR2 (顺) R1CH=CHR2 (反) R1R2CH=CHR3 面外弯曲振动位置/cm-1 995 ~985, 910 ~905 895 ~ 885 730 ~ 650 980 ~ 965 840 ~ 790
第三章 红外吸收光谱分析-1

波长和波数
红外区光谱用波长和波数( 红外区光谱用波长和波数(wave number) 波长和波数 ) 来表征 ; 波长多用m做单位; 做单位; 波长多用 做单位 波数: 表示, 波数:以σ表示,定义为波长的倒数,单位 表示 定义为波长的倒数, cm-1,其物理意义是每厘米长光波中波的数 目. σ=1/λ(cm)=104/λ(m)=υ/c 用波数表示频率的好处是比用频率要方便, 用波数表示频率的好处是比用频率要方便,且 数值小. 数值小. 一般用透光率 波数曲线或透光度-波长曲线 透光率-波数曲线 波长曲线来 一般用透光率 波数曲线或透光度 波长曲线来 描述红外吸收光谱. 描述红外吸收光谱.
第三章 红外吸收光谱分析
3.2 基本原理 3.2.1 产生红外吸收的条件
产生红外吸收的条件
1) 辐射光子具有的能量与发生振动 跃迁所需的跃迁能量相等. 跃迁所需的跃迁能量相等. 2)辐射与物质之间有耦合作用. )辐射与物质之间有耦合作用.
条件一: 条件一:辐射光子的能量应与振动跃 迁所需能量相等
红外光谱的特点-1 红外光谱的特点
紫外,可见吸收光谱常用于研究不饱和 紫外,可见吸收光谱常用于研究不饱和 有机物, 有机物,特别是具有共轭体系的有机化 合物; 红外光谱法主要研究在振动中 合物;而红外光谱法主要研究在振动中 伴随有偶极矩变化的化合物. 伴随有偶极矩变化的化合物. 因此,除了单原子和同核分子如Ne, , 因此,除了单原子和同核分子如 ,He, O2,H2等之外,几乎所有的有机化合物 等之外, 在红外光谱区均有吸收. 在红外光谱区均有吸收. 一般只要结构上不同, 一般只要结构上不同,就会有不同的红 外光谱图. 外光谱图.
红外光谱的特点-2 红外光谱的特点
红外谱图吸收带的位置与吸收谱带的强 红外谱图吸收带的位置与吸收谱带的强 度反映了分子结构上的特点, 度反映了分子结构上的特点,可以用来 定基团,定结构; 定基团,定结构; 谱带的强度与分子组成以及含量有关 与分子组成以及含量有关, 谱带的强度与分子组成以及含量有关, 可以用来进行定量分析及纯度的检查; 可以用来进行定量分析及纯度的检查; 红外光谱分析特征性强,气体, 红外光谱分析特征性强,气体,液体和 固体样品均可以测定,并且具有用量少, 固体样品均可以测定,并且具有用量少, 分析速度快和不破坏样品等特点. 分析速度快和不破坏样品等特点.
第三章红外光谱IR
烷烃吸收峰
正己烷的红外光谱图
2,2,4-三甲基戊烷的红外光谱图
2、不饱和烃
• 烯烃 • 炔烃 • 芳香烃
2、1 烯烃 烯烃双键的特征吸收
影响双键碳碳伸缩振动吸收的因素
• 对称性:对称性越高,吸收强度越低。 • 与吸电子基团相连,振动波数下降,吸
收强度增加。 • 取代基的质量效应:双键上的氢被氘取
代后,波数下降10-20厘米-1。质量效应 • 共轭效应:使波数下降约30厘米-1 。
1-己烯的红外光谱图
~3060cm-1: 烯烃C—H伸缩振动;~1820:910cm-1倍频; ~1650cm-1: C=C伸缩振动;~995,905cm-1: C=CH2 非平面摇摆振动
顺式和反式2,2,5,5-四甲基己烯红外光谱 a 顺式 b 反式
v~
=
1
——
K
2C M
M = m1 m2 m1 + m2
双原子分子红外吸收的频率决定于折合质量和键力常数。
C-H C-C C-O C-Cl C-Br C-I
-1 cm
3000
1200 1100
800
550
500
v cm-1
力常数/g.s-2
CC 2200~2100
12~18105
C=C 1680~1620
C-H面外弯曲振动吸收峰位置(cm-1) 670
770-730,710-690 770-735
810-750,710-690 833-810
780-760,745-705 885-870,825-805 865-810,730-675
810-800 850-840 870-855
870
各类取代苯的倍频吸收和面外弯曲振动吸收
有机波谱解析-第三章_红外光谱

由于红外光谱吸收强度受狭缝宽度、温度和溶剂等因素影 响,故不易精确测定,在实际分析中,只是通过与羰基等强吸 收峰对比来定性研究。
谱带强度与振动时偶极矩变化有关,偶极矩变化愈 基团极性 大,谱带强度愈大;偶极矩不发生变化,谱带强度为0, 即为红外非活性。 电子效应
红外吸收强度 偶极距变化幅度 振动偶合
伸缩振动(
as
)两种形式。
弯曲振动:原子垂直于化学键方向的运动。又可以分
它们还可以细分为摇摆、卷曲等振动形式。
为面内弯曲振动()和面外弯曲振动( )两种形式,
+和-表示垂直于纸面方向的前后振动。
亚甲基的振动形式
三、分子振动与红外吸收峰的关系
理论上具有特定频率的每一种振动都能吸收相应 频率的红外光,在光谱图对应位臵上出现一个吸收 峰。实际上,因种种原因分子振动的数目与谱图中
纵坐标为: 百分透过率(%) 横坐标为: 波长(µ m)或波 数(cm-1)。
环戊烷
也可用文字形式表示为:2955cm-1(s)为CH2的反对称伸缩振动 (υasCH2),2870cm-1(m)为CH2的对称伸缩振动(υsCH2) 1458cm-1(m) 为CH2的面内弯曲振动(δ面内CH2),895cm-1(m)为CH2的面外弯曲振动 (面外CH2)
诱导效应大于共轭效应, C=O 蓝移至 1735 cm-1
三、空间效应
(1)空间位阻 破坏共轭体系的共平面性,使共
轭效应减弱,双键的振动频率蓝移(增大)。
CH(CH3)2 O O O
CH3 CH3
CH3 CH(CH3)2
CH3
1663cm-1
1686cm-1
1693cm-1
(2)环的张力:环的大小影响环上有关基 团的频率。
第三章 红外吸收光谱法

红外吸收光谱法
01 基础知识
02 光栅型红外分光光度计
目录
CONTENTS
03 傅里叶变换红外光谱仪 04 样品的制备
05 实训五 乙酰苯胺的红外光谱测定
06 实训六 苯乙酮的红外光谱测定
案例 导入
药检中经常会遇到硫酸小诺霉素注射液与硫酸庆 大霉素注射液,虽然两者临床药理作用和毒副反应 相差甚多,但从它们的显色反应、薄层斑点位置等 化学鉴定方法来看,两者是难以进行区分的,由于 硫酸小诺霉素注射液在市场上的出售价格要比硫酸 庆大霉素注射液高出许多倍,这样就导致一些不法 分子利用这可乘之机,来进行假药的制作与销售。 因此,必须严把药品质量关,解决这一问题。用什 么方法可以高度准确地将问题彻底解决呢?
04 典型光谱
1 . 芳烃类 取代苯的主要特征峰有: νΦ—H3100~3030cm-1(m);νC=C(骨架振动)~1600cm-1(m或s)及~ 1500cm-1(m或s);γΦ—H910~665cm-1(s);泛频峰2000~1667cm-1(w,vw)。现 以甲苯为例说明取代苯的红外吸收特征,如图3-1所示。
04 典型光谱
2 . 醇、酚、羧酸类 (3)νC=O
νC=O是此三类化合物中羧酸独有的重要特征吸收峰,峰位为1740~1650cm-1的高 强吸收峰,干扰较少。可据此区别羧酸与醇和酚。
04 典型光谱
3 . 醛、酮类 (1)醛类
主要特征峰:νC=O1725cm-1(s)及醛基氢νO=C—H~2820与2720cm-1两个吸收 峰。若羰基与双键或芳环共轭,将使νC=O峰向低波数方向移动至1710~1685cm-1。
2 . 醇、酚、羧酸类
图 3-4 正辛醇、丙酸、苯酚的红外吸收光谱图
第三章-红外吸收光谱分析

第三章红外吸收光谱分析3.1概述3.1.1红外吸收光谱的基本原理红外吸收光谱法又称为分子振动转动光谱,属于分子光谱的范畴,是有机物结构分析的重要方法之一。
当一定频率的红外光照射分子时,若分子中某个基团的振动频率和红外辐射的频率一致,两者产生共振,光的能量通过分子偶极矩的变化传递给分子,该基团就吸收了这个频率的红外光,产生振动能级跃迁;如果红外辐射的频率和分子中各基团的振动能级不一致,该频率的红外光将不被吸收。
如果用频率连续变化的红外光照射某试样,分子将吸收某些频率的辐射,引起对应区域辐射强度的减弱,用仪器以吸收曲线的形式记录下来,就得到该试样的红外吸收光谱,稀溶液谱带的吸光度遵守Lambert-Beer定律。
图3-1为正辛烷的红外吸收光谱。
红外谱图中的纵坐标为吸收强度,通常用透过率或吸光度表示,横坐标以波数或波长表示,两者互为倒数。
图中的各个吸收谱带表示相应基团的振动频率。
各种化合物分子结构不同,分子中各个基团的振动频率不同。
其红外吸收光谱也不同,利用这一特性,可进行有机化合物的结构分析、定性鉴定和定量分析。
图3-1 正辛烷的红外光谱图几乎所有的有机和无机化合物在红外光谱区均有吸收。
除光学异构体,某些高分子量的高聚物以及一些同系物外,结构不同的两个化合物,它们的红外光谱一定不会相同。
吸收谱带出现的频率位置是由分子振动能级决定,可以用经典力学(牛顿力学)的简正振动理论来说明。
吸收谱带的强度则主要取决于振动过程中偶极矩的变化和能级跃迁的概率。
也就是说,红外光谱中,吸收谱带的位置、形状和强度反映了分子结构的特点,而吸收谱带的吸收强度和分子组成或官能团的含量有关。
因此,红外吸收光谱在化学领域中的应用,大体上可分为两个方面,即分子结构的基础研究和用于化学组成的分析。
首先,红外光谱可以研究分子的结构和化学键。
利用红外光谱法测定分子的键长和键角,以此推断出分子的立体构型;利用红外光谱法测定分子的力常数和分子对称性等,根据所得的力常数就可以知道化学键的强弱;由简正频率来计算热力学函数等等。
材料分析方法第三章_红外光谱剖析

烯烃类型对=C-H的面外弯曲振动的影响
对判断烯烃类 型非常有用
烯烃类型
R1CH=CH2 R1R2C=CH2 R1CH=CHR2(顺) R1CH=CHR2(反) R1R2C=CHR3
(4)分析时间短。一般红外光谱做一个样可在10~30分钟 内完成。如果采用傅里叶变换红外光谱仪在一秒钟以内 就可完成扫描。为快速分析的动力学研究提供了十分有 用的工具。
(5)所需样品用量少,且可以回收。红外光谱分析一次 用样量约1~5mg,有时甚至可以只用几十微克。
IR光谱表示法: 横坐标为吸收波长(m),或吸收频率(波数
1947年第一台实用的双光束自动记录的红外分光光度计 问世。这是一台以棱镜作为色散元件的第一代红外分光光 度计。
到了六十年代,用光栅代替棱镜作分光器 的第二 代红外光谱仪投入了使用。这种计算机化的光栅为分光 部件的第二代红外分光光度计仍在应用。
七十年代后期,干涉型傅里叶变换红外光谱仪(FTIR)投入了使用,这就是第三代红外分光光度计。
断烯烃的存在 , 苯环 C-H 大于3000
初步判断烯烃结构 的存在
影响碳碳双键伸缩振动吸收的因素
对称性:对称性越高,吸收强度越低。 取代基:与吸电子基团相连,振动波数下
降。 取代基的质量效应:双键上的氢被氘取代
后,波数下降10-20 厘米-1。 共轭效应:使波数下降约30厘米-1 。
1-己烯的红外光谱图
吸收池和检测器
由于玻璃,石英等常规透明材料不能透过红外线, 因此红外吸收池必须采用特殊的透红外材料制作 如:NaCl,KBr,CsI,KRS-5等作为窗口。由于 该类材料均属于无机盐,很容易吸收水汽发生潮 解。固体粉体样品可以直接与KBr混合压片,直 接进行测定。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
3-6-2 不饱和度的计算
不饱和度定义:当一个化合物衍变为相应的烃后,与其 同碳的饱和开链烃比较,每缺少两个氢则为一个不饱和 度。因此,分子的不饱和度双键为1,三键为2,一个脂 环为1,一个苯环为4。
计算不饱和度的经验公式:U=1+2n6+n4+(3n5+n3-n1)/2 n6,n5,n4,n3,n1分别为VI价,V价,IV价,III价,I价的原子数
例3解:
1)不饱和度 分子式: C8H8,
2)峰归属
U=1-8/2+8=5
波数(cm-1 )
归属
结构信息
3100~3000cm-1 苯环和双键(=C-H)伸缩振动, ν (=C-H) =C-H,Ar-H
1630 cm-1
1600 ,1580, 1500,1450
990,910 770,700
C=C伸缩振动峰, ν (C=C) 芳环C=C伸缩振动,ν (C=C)
第三章 红外吸收光谱
Infrared Spectra
3-1 红外光谱的基本知识
3-1-1 红外光的区域
0.75mm
175m1 (cm)
3-1-2 红外光谱的表示方法
仲丁基苯的红外吸收光谱
吸光度: A = log1/T = logI/Io
>C=CH2
3)官能团
>C=CH2
OH
CH2
CH3
4)确定结构
CH2
C CH2OH CH3
例6.化合物C4H8O,根据如下IR谱图确定结构, 并说明依据。
例6解:
1)不饱和度 分子式: C4H8O, U=1-8/2+4=1 2)峰归属
(2) 弯曲振动 d (bending
vibration)
(a) 对称伸缩 s
(b) 不对称伸缩 as
(a) 平面摇摆 (b) 剪式振动 (c) 非平面摇摆 (d) 扭曲振动
面内弯曲振动 dip
面外弯曲振动 g/doop
3-2-2 分子振动的经典力学模型
双原子分子 的振动频率:
1
2
m m1m2
例2解:
1)不饱和度 分子式: C4H8O2,
2)峰归属
U=1-8/2+4=1
波数(cm-1 )
归属
结构信息
3000~2800cm-1
1740 cm-1 1460cm-1 1380cm-1 1249cm-1 1049cm-1
是-CH2或-CH3的C-H伸缩振动峰,ν (CH3)和 ν (CH2) 强峰,C=O伸缩振动峰, ν (C=O)
1170
C-C伸缩振动吸收峰。 ν (C-C)
817
苯环上=C-H的面外变形振动。
结构信息 C=C-H CH3
C≣N 苯环
CH3特征
苯环对位取代
3)官能团 CH3
C≡N
4)确定结构
H3C
苯环
CN
6)验证结构 不饱和度与计算结果相符;并与标准谱图对照证明结构正确。
例2.某化合物分子式C4H8O2,试根据如下红外 光谱图,推测其结构。
p-共轭体系要同时考虑到+C效应和-I效应。
(3)空间效应
空间障碍造成羰基与双键的共平面性降低,共轭 效应受到限制,振动频率向接近正常值的方向移动。
(4)环张力效应之一
环变小时,成环的弯曲C-C键成键强度降低,由于键 角地减小,环内C=C双键的双键性迅速降低,力常数减小, 伸缩振动的频率随之下降。
E = h = hc/
透过率: T% = I/Io x 100%
3-1-3 红外光谱的常用术语
基频峰:振动能级从基态跃迁到第一激发态时所产生的吸收峰; 泛频峰:包括倍频峰和组频峰(分为合频峰和差频峰); 特征区:4000~1330cm-1(2.5~7.5mm)之间的高频区; 指纹区:1330~400cm-1(7.5~25mm)之间的低频区; 特征峰:特征区中能用于鉴定原子基团的吸收峰; 相关峰:与特征峰相互依存而又相互可以佐证的吸收峰; 费米共振:泛频峰接近基频峰时,发生相互作用,形成为比原
1)根据分子式,计算未知物的不饱和度U,估计分子 结构中是否有双键、三键或芳香环; 2)根据未知物的红外光谱,找出主要的强吸收峰,按 照由简单到复杂的顺序,分五个区来分析: 4000~2500cm-1: X-H(X=C.N.O.S)的伸缩振动区 2500~2000cm-1: 三键和累积双键(-C≡C-,-C ≡ N,
3068cm-1 3044cm-1
(2) (3)
2943cm-1 1765 cm-1
谱峰归属 (4) (5) (6) (7) (8)
推测结构
1594 cm-1 1493 cm-1
1371 cm-1
1194 cm-1
1027 cm-1
750cm-1 7692cm-1
可能含有苯环和含有C=O、C=C或环。 苯环上=C-H伸缩振动,说明可能是芳香族化合物。
k
m1 m2
m k: 2个原子又平衡位置伸 长0.1nm后的回复力
3-2-3 红外光谱的特点
分子的振动能级跃迁吸收发生在红外区; 分子只吸收与其振动频率相同的光子; 分子振动的频率决定于成键原子的质量和键
的强度; 相同基团有其固定的特征红外吸收区域(可
能不只一处); 不同基团的特征吸收区及吸收强度不同。
环外双键的伸缩振动频率如何变化?
为什么?
(4)环张力效应之二
理论上,C=O双键中的C是sp2杂化态,但是在环变小 时,由于成环的弯曲C-C键需要较大的p电子成分,相应 地,环外C=O双键的C原子p电子成分较小,s电子成分变 大,力常数增加,伸缩振动需要更高地能量。
(5)氢键效应
CH3 C CH2 C CH3
例7:醇类化合物
例8:酚类化合物
例9:醚类化合物
例10:羰基化合物-醛类
例11:羰基化合物-酮类
例12:羧酸类化合物
例13:酯类化合物
例14:酸酐类化合物
例15:酰卤类化合物
例16:酰胺类化合物
例17:胺类化合物
例18:腈类化合物
3-6 红外光谱图的解析
3-6-1 波谱解析的一般步骤
羰基上连有强吸电子基时,C=O双键的电子云向碳偏移, 从而使C=O的双键性增加,两个原子间的力常数k增大, C=O的伸缩振动的频率向高波数移动。
(2)共轭效应
羰基上连有双键或带孤电子对的杂原子时,可以形成共轭或p-共轭效应。 -共轭体系中单双键的键长平均 化、电子云密度平均化,从而使C=O的双键性减小,力常 数k降低,C=O的伸缩振动的频率向低波数移动。
C=O
C-O
O
H C O CH2CH2CH3 (a)
ν as(C-O-C) 1180cm-1
O
O
H3C C O CH2CH3 (b)
1240cm-1
H3CH2C C O CH3 (c)
1160cm-1
5)确定结构
O
H3C C O CH2CH3
例3.某化合物分子式C8H8,试根据如下红外光 谱图,推测其结构。
O
非对称性伸缩振动
asC=O 1828cm-1
问题:
孤立的-CH3的C-H对称弯曲振动在 1380cm-1 (单峰)附近,但是异丙基-CH(CH3)2 中的甲基C-H的弯曲振动有两个吸收峰,分 别在~1385cm-1 和~1370cm-1。为什么?
(7)费米共振
HO C
dC-H
OCH3
醛基的C-H键的弯曲振动在1390cm-1附近,其倍频吸收 和醛基C-H键的伸缩振动区域2850~2700cm-1十分接近,两 者发生费米共振。红外吸收峰偏离基频值。
如果化合物不含V价以上的元素,则公式简化为: U=1+n4+(n3-n1)/2
注意:(1)II价元素不参与计算;(2) 元素化合价按分子 中原子实际提供的成键电子数计算;(3)元素化合价不 分正负,只按价数分类;(4)若分子只含变价元素,计 算多种可能性,然后根据光谱数据取舍。
3-6-3 解析的经验程序
甲基不对称变形振动峰和CH2剪式振动的迭合。 CH3的C-H对称变形振动,δ (CH3)甲基特征。 C-O-C不对称伸缩振动峰, ν as(C-O-C)
C-O-C对称伸缩振动峰。 ν s(C-O-C)
CH3, CH2
酯C=O CH2,CH3 CH3 酯的特征 酯的特征
3)官能团
CH2
CH3
4)可能的结构
饱和碳氢C-H伸缩振动。 C=O伸缩振动吸收峰,波数较高,可能为苯酯(=CO-COR,~1760 cm-1)。 芳环C=C骨架伸缩振动。没有裂分,说明没有共轭基 团与苯环直接相连。 CH3的对称变形振动,波数低移,可能与羰基相连。 C-O-C不对称伸缩振动,酯的特征。 C-O-C对称伸缩振动。 苯环上相邻5个H原子=C-H的面外变形振动和环骨架变 形振动,苯环单取代的特征。
-C=C=C-,-N=C=O,-N=C=S等)的伸缩振动区 2000~1500cm-1: 双键的伸缩振动区,如 C=C,C=O,
N=C,N=O,苯环骨架等 1500~1300cm-1: 主要提供C-H弯曲振动的信息 1300~400cm-1: 单键的伸缩振动、分子骨架振动、双
键上C-H面外弯曲振动信息
归属
3030
苯环上=C-H伸缩振动, ν (=C-H)
~2920 2217
是-CH2或-CH3的C-H非对称伸缩振动峰,此峰很 弱,可能是芳环上连的烃基。 ν (CH3)和 ν (CH2) CN的特征吸收峰, ν (C≣N)
1607,1508 芳环C=C伸缩振动,ν (C=C)
1450 1384
芳环C=C伸缩振动和CH3的C-H变形振动的迭合。 CH3的C-H对称变形振动δ (CH3)。