药物Copanlisib(库潘尼西)合成检索总结报告
药物Abemaciclib(玻玛西林)合成检索总结报告
药物Abemaciclib(玻玛西林)合成检索总结报告一、Abemaciclib(玻玛西林)简介Abemaciclib(玻玛西林)于2017年9月在美国上市,主要用于晚期或复发性乳腺癌的治疗。
Abemaciclib(玻玛西林)常见的不良反应有腹泻、肝毒性、静脉血栓栓塞等等。
Abemaciclib(玻玛西林)分子结构式如下:CAS:1231929-97-7英文名称:Abemaciclib中文名称:玻玛西林本文主要对Abemaciclib(玻玛西林)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
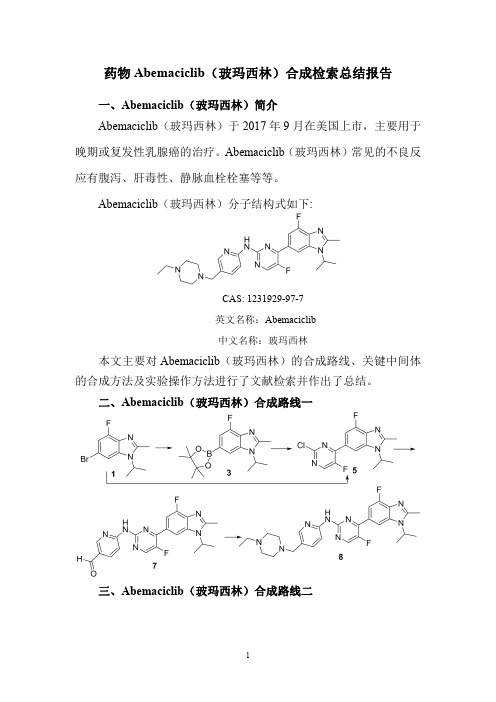
二、Abemaciclib(玻玛西林)合成路线一三、Abemaciclib(玻玛西林)合成路线二四、Abemaciclib (玻玛西林)合成路线一检索总结报告(一)Abemaciclib (玻玛西林)中间体3的合成(路线一)合成方法实验步骤参考文献操作方法一DMSO (100mL)was added to a 500mL one-necked flask and the compound of formula 1(20.0g,73.80mmoL),bis (pinacolato)diboron 2(27.6g,108.69mmol),tricyclohexylphosphine (3.53g,12.61mmol),Potassium acetate (21.3g,217.38mmol).Nitrogen gas was added to the reaction flask after the rapid addition of palladium acetate (1.5g),nitrogen PaulProtect the next open heating to 90o C.After 3hours the reaction flask was cooled to room temperature and the reaction mixture was poured into 700mL of water,mixed thoroughly and subtracted PressureCN107266421;(2017);(A)Chinesefiltration,the filter cake was washed twice with100mL of water and dried to give a light brown solid.Crude was added 50mL petroleum ether,10mL Ethyl acetate beating10min, vacuum filtration to give a white solid3(18.8g,yield80.3%)操作方法二Under nitrogen protection,palladium acetate(28mg)and tricyclohexylphosphine(54.3mg)was added into thesolution of6-bromo-4-fluoro-1-isopropyl-2-methyl-H-benzo[d]imidazole1(300mg,1.10mmol),bis(pinacolato)-diboron2(422mg,1.70mmol)and potassium acetate(326mg,3.3mmol)in anhydrous dimethyl sulfoxide(DMSO,5mL),and the reaction was carried out at100°C.undernitrogen protection for2hours.After cooling to roomtemperature,the reaction was filtered on Celite,the filtercake was washed with ethyl acetate,the filtrate was washedwith brine,dried over anhydrous sodium sulfate,the filtratewas concentrated under reduced pressure,and the filtrate wasseparated on column chromatography(eluant:petroleumether/ethyl acetate(v/v)=3:1),to affard260mg of a paleyellow solid3.US2019/152954;(2019);(A1)English操作方法三A suspension of6-bromo-4-fluoro-2-methyl-1-(propan-2-yl)-1H-benzimidazole(1)(90g,331.95mmol),bis(pinacolato)-diboron(2)(126g,498mmol),AcOK(80g,815.15mmol), tricyclohexylphosphine(14g,49.8mmol),and Pd(OAc)2(7.45g,33.2mmol)in DMSO(1.0L)was sparged with N2and then stirred at100°C.for16h.TLC analysis(1:1petroleum ether/EtOAc)showed complete consumption ofthe starting material.The black suspension was poured intoH2O(3.0L)and extracted with EtOAc(2*3L).Thecombined organic phases were washed with brine,dried overNa2SO4,filtered,and concentrated.The residue was purifiedby flash chromatography(Biotage,1.0kg,0-40%EtOAc/petroleum ether)to provide4-fluoro-2-methyl-1-(propan-2-yl)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-benzimidazole(3)(77g,73%yield)as a yellow solid.US2019/330196;(2019);(A1)English操作方法四The6-Bromo-4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole1(9.0g,33.2mmol),Bis-boronic acid pinacolester2(0.65g,49.8mmo1),palladium acetate(840mg), tricyclohexylphosphine(1.63g)and potassium acetate(9,78g,99.8mmo1)were added to60mL of DMSO and thenitrogen was allowed to warm to80°C for6h.The organicphase was extracted with EA,and the organic phase waswashed with EA.The organic phase was dried withanhydrous sodium sulfate and filtered through the filtrate.The filtrate was concentrated and purified by silica gelCN104910137;(2017);(B)Chinese;EP3091008;(2016);(A1)English。
新药Opaganib(奥帕尼布)合成检索总结报告

1新药Opaganib (奥帕尼布)合成检索总结报告
一、Opaganib (奥帕尼布)简介
Opaganib (奥帕尼布)的适应症有抗癌、抗病毒和抗炎。
2020年4月17日官方宣布,向美国食品和药物管理局提交了新药申请,研究用Opaganib (奥帕尼布)治疗新型冠状病毒肺炎COVID-19。
该药曾从美国FDA 获得了用于治疗胆管癌的孤儿药资格,并且正在晚期胆管癌的1/2a 期研究和前列腺癌的2期研究中,并且正在评估已确诊的以色列COVID-19患者中冠状病毒(COVID-19)的疗效。
Opaganib (奥帕尼布)分子结构式如下
:
CAS :915385-81-8
英文名称:Opaganib
中文名称:奥帕尼布
本文主要对Opaganib (奥帕尼布)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Opaganib
(奥帕尼布)合成路线
三、Opaganib (奥帕尼布)合成检索总结报告
(一)Opaganib (奥帕尼布)中间体2
的合成合成方法实验步骤
参考文献A two-neck round bottom flask was connected to a reflux condenser,to which aluminum chloride (AlCl 3,7.15mmol)was injected,followed by cooling at -5°C.in argon。
药物帕布昔利布(palbociclib)合成检索总结报告
药物帕布昔利布(palbociclib)合成检索总结报告一、帕布昔利布(palbociclib)简介帕布昔利布(palbociclib)于2015年2月在美国上市,主要用于治疗绝经后妇女晚期乳腺癌。
帕布昔利布(palbociclib)常见的不良反应有食欲下降、呕吐、无力、脱发等等。
帕布昔利布(palbociclib)分子结构式如下:英文名称:palbociclib中文名称:帕布昔利布本文主要对帕布昔利布(palbociclib)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
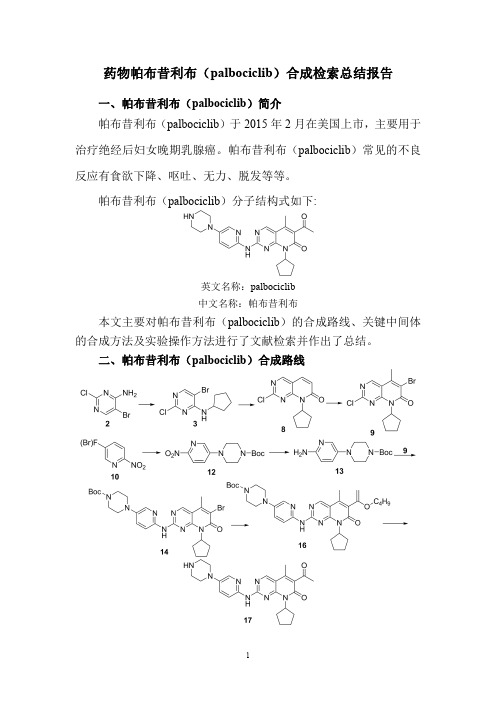
二、帕布昔利布(palbociclib)合成路线三、帕布昔利布(palbociclib )合成检索总结报告(一)帕布昔利布(palbociclib )中间体3的合成方法一合成方法实验步骤参考文献操作方法一Compound 2(208.4g,1mol)was added to the 2L three-necked flask,Copper trifluoroacetate (2.8g,0.01mol)1,4-dioxane (833.6g),Sodium hydroxide (60g,1.5mol),cyclopentyl bromide 1(223.6g,1.5mol),incubate at 80o C for 6hours,the reaction is over,Add saturated ammonium chloride solution (832g)and stir well,extract with ethyl acetate (300g),the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure,the obtained solid was collected in a 2L three-necked flask,and isopropyl alcohol (832g)was added,activated carbon (10g),stirred at 60°C for 1hour,filtered while hot,The filtrate was naturally cooled to 10°C to precipitate solids and filtered,The filter cake was vacuum dried at 40°C to obtain Compound 3,a white solid 249g,and a yield of 90%.The HPLC purity was 99.5%.CN108299311;(2018);(A)Chinese(二)帕布昔利布(palbociclib )中间体3的合成方法二合成方法实验步骤参考文献操作方法一Compound 2(208.4g,1mol)was added to the 2L three-necked flask,Copper acetate (2g,0.01mol),dichloroethane (625g),sodium hydroxide (100g,2.5mol),cyclopentyl chloride 4(261.5g,2.5mol),incubate at 80o C for 10hours,After the reaction,add saturated ammonium chloride solution (832g)and stir well.Extracted with ethyl acetate (300g),the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure,The obtained solid was collected in a 2L three-necked flask,ethyl acetate (832g)was added,activated carbon (10g),stirred at 60°C for 1hour,filtered while hot,The filtrate was naturally cooled to 10°C to precipitate solids and filtered,The filter cake was vacuum dried at 40°C to obtain Compound 3,white solid 213g,yield 77%,The HPLC purity was 98.5%.CN108299311;(2018);(A)Chinese(三)帕布昔利布(palbociclib )中间体3的合成方法三合成方法实验步骤参考文献操作方法一To a solution of 5-bromo-2,4-dichloropyrimidine 6(45.6g,200mmol)in dioxane (400mL)was added N-cyclopentylamine 5(20.4g,240mmol)at room temperature.The mixture thus obtained was stirred at room temperature for 6h.The reaction mixture was then diluted with ethyl acetate and washed with brine and dried over MgSO 4.The solvent was evaporated to give the title compound as a light yellow solid 3(56g,100%)which was used in next step without further purification.WO2009/85185;(2009);(A1)English 操作方法二2,4-dichloro-5-bromopyrimidine 6(200g,888mmol)and 1.4L of dichloromethane were added to a reaction flask,and sodium hydrogencarbonate (372g,4440mmol)was added.At room temperature,cyclopentylamine 5(90.7g,1066mmol)diluted with 0.6L of dichloromethane was slowly added dropwise,and reacted at room temperature for 13h.The sodium hydrogencarbonate and the resulting inorganic salt were removed by suction filtration,the mother liquor was evaporated to remove dichloromethane,and 1.0L of n-heptane was added to recrystallize.236.7g of a white solid were obtained,the yield of 3was 97.5%,the purity was >99.8%,and the impurity 5-bromo-4-chloro-2-cyclopentyl-Aminopyrimidine.Melting point:94-96°109206373;(2019);(A)Chinese 操作方法三5-Bromo-2,4-dichloropyrimidine 6(227.9g,1mol)was sequentially added to a 2L three-necked flask.Isopropyl alcohol (360g,6mol),diisopropylethylamine (167.7g,1.3mol)was added at -15°C.A solution of cyclopentylamine 5(106.2g,1.3mol)in isopropanol (120g,2mol)was added dropwise.The reaction was carried out for 2to 3hours,and the solid was filtered.The filter cake was taken in a 2L reaction flask,petroleum ether (600g)was added thereto,stirred for 1hour,filtered again,and the filter cake was washed with petroleum ether (100g).The filter cake was collected and dried under vacuum at 40°C to give 260.0g of Intermediate II,white solid,yield 94%.CN108299422;(2018);(A)Chinese A solution of 5-bromo-2,4-dichloropyrimidine (6)(5g,22mmol)and N,N-diisopropylethylamine (5.7g,44mmol)in ethanol (30mL)was cooled to 10°C under nitrogenCombinatorial Chemistry and操作方法四atmosphere.Charged cyclopentylamine 5(2.1g,24.1mmol)and stirred for 5hours at 40°C.Progress of the reaction wasmonitored by TLC and solvent was evaporated undervacuum.Resulting residue was stirred with hexane (20mL)for 2hours at 0°C.Precipitate was filtered and washed withhexane to obtain compound (3).Off-white crystalline solid,Yield:90%,mp.95-97°C.High Throughput Screening ;vol.20;nb.8;(2017);p.703–712.操作方法五In a large sealed tube is added 5-bromo-2,4-dichloropyri-midine 6(3g,13.2mmol)in 100mL of EtOH.Then cyclopentyl amine 5(1.95mL,19.75mmol)and N,N'-diisopropylethylamine (3.36mL,19.8mmol)are added to the solution at rt.The solution is then stirred rt overnight.Solvent is evaporated and the crude is purified using silica gel chromatography (15%ethyl acetate/85%hexane)to give (5-bromo-2-chloro-pyrimidin-4-yl)-cyclopentyl-amine 3as a white solid (3.25g,89%).WO2010/20675;(2010);(A1)English 操作方法六To a vessel was added absolute ethanol (3000mL,3.0vol)followed by 5-bromo-2,4-dichloropyrimidine 6(mw 227.87;1000g,1.0equiv.).Triethylamine (612mL,1.0equiv.)was added,and then cyclopentylamine 5(mw 85.15;520mL,1.2equiv.)was added slowly over 2hours to control the mild exotherm.After completion of cyclopentylamine addition,the reaction was seeded with 5-bromo-2-chloro-6-cyclopentylamino-pyrimidine 3(5g,0.5wt%)to induce crystallization,if needed.The reaction was stirred at 25°C for 2hours.Water (2500mL,2.5vol)was added to the vessel at 20-25°C at a rate of 30mL/min.The mixture was cooled to 8-12°C at 2°C/min.The slurry was kept at 8-12°C for 1hour and then filtered onto a 2Whatman paper filter.The cake was rinsed with n-heptane (2000mL).The cake was reslurried with n-heptane on the filter drier (2000mL).The material was dried overnight in the vacuum oven at 50-55°C to give 5-bromo-2-chloro-6-cyclopentylamino-pyrimidine 3(1020g;84%)as a white solid.WO2014/128588;(2014);(A1)English(四)帕布昔利布(palbociclib )中间体8的合成合成方法实验步骤参考文献To a vessel was added 5-bromo-2-chloro-6-cyclopentyl-amino-pyridimidine 3(10.0g,1.0equiv.)along with。
芦曲泊帕合成检索总结报告-概述说明以及解释

芦曲泊帕合成检索总结报告-概述说明以及解释1.引言概述部分是对本篇长文的一个简要介绍,可以包括对芦曲泊帕合成检索总结报告的背景和主题的概括。
以下是一个概述部分的示例内容:1.1 概述芦曲泊帕(Lupuqupa) 是一种常见的化学物质,具有广泛的应用领域。
随着科学技术的快速发展和人们对新药物的迫切需求,芦曲泊帕作为一种重要的抗癌药物,受到了广泛的研究和关注。
在本篇文章中,我们对芦曲泊帕的合成方法和检索技术进行了综合总结和归纳。
本文旨在系统地介绍芦曲泊帕的定义、背景及其相关研究领域的最新进展。
首先,我们将给出对芦曲泊帕的详细定义,并介绍其在医药领域中的重要性和应用价值。
随后,我们将深入探讨芦曲泊帕的合成方法,包括传统合成路线和近期新兴的绿色合成方法。
此外,我们还将介绍芦曲泊帕的检索方法,包括文献检索、数据库检索和分子结构检索等。
本报告的目的是为读者提供一个清晰而全面的了解芦曲泊帕合成和检索方面的最新研究进展。
通过对相关文献的综合分析和总结,我们将对芦曲泊帕的合成和检索方法进行归纳,提出一些建议和展望未来的研究方向。
我们相信,本篇长文将对化学研究者和医药科学家在芦曲泊帕的研究和应用方面提供有价值的参考和指导。
(注意:以上内容仅为示例,请根据具体情况进行修改和完善。
)文章结构部分的内容如下:1.2 文章结构本篇长文主要分为引言、正文和结论三个部分。
引言部分主要介绍了文章的概述、文章结构和文章的目的。
在概述部分,将简要介绍芦曲泊帕的背景和相关信息,引起读者的兴趣。
接着,在文章结构部分,将详细说明文章的组织结构和各个部分的内容,为读者提供对整篇文章的整体把握。
最后,在目的部分将明确本篇文章的写作目的和预期的研究成果,为后续的内容做铺垫。
正文部分是本文的核心部分,主要分为芦曲泊帕的定义和背景、合成方法以及检索方法三个部分。
在芦曲泊帕的定义和背景部分,将全面介绍芦曲泊帕的基本信息、性质特点以及其在相关领域的应用。
奥拉帕尼(Olaparib)合成检索总结报告
奥拉帕尼(Olaparib)合成检索总结报告
一、奥拉帕尼(Olaparib)简介
奥拉帕尼(Olaparib)于2014年12月19日在美国上市。
奥拉帕尼(Olaparib)是聚腺苷酸二磷酸核糖转移酶抑制剂,用于晚期卵巢癌。
奥拉帕尼(Olaparib)不良反应有:恶心、疲乏、呕吐、腹泻、味觉障碍、消化不良等等。
奥拉帕尼(Olaparib)分子结构式如下:
英文名称:Olaparib
中文名称:奥拉帕尼
本文主要对奥拉帕尼(Olaparib)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
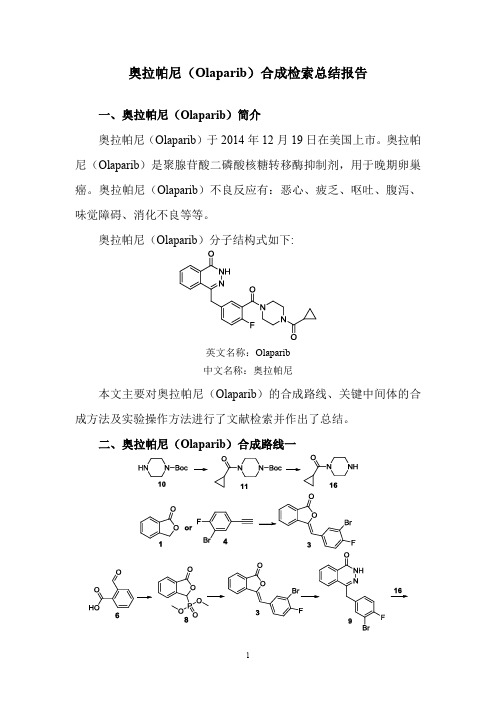
二、奥拉帕尼(Olaparib)合成路线一
三、奥拉帕尼(Olaparib)合成路线二
四、奥拉帕尼(Olaparib)合成路线一检索总结报告
(一) 奥拉帕尼(Olaparib)中间体3的合成方法一(路线一)
(二) 奥拉帕尼(Olaparib)中间体3的合成方法二(路线一)
(三) 奥拉帕尼(Olaparib)中间体3的合成方法三(路线一)①奥拉帕尼(Olaparib)中间体8的合成。
新药Capmatinib(卡马替尼)合成检索总结报告
新药Capmatinib(卡马替尼)合成检索总结报告一、Capmatinib(卡马替尼)简介Capmatinib(卡马替尼)于2020年5月6日获得美国食品药品监督管理局(FDA)批准上市,用于治疗MET外显子14跳跃突变的转移性非小细胞肺癌(NSCLC)成年患者。
同时,FDA批准了检测该类基因突变的产品。
Capmatinib曾获得FDA授予的突破性疗法称号。
2020年2月11日,诺华宣布FDA接受了Capmatinib的新药申请并给予了优先审查。
Capmatinib(卡马替尼)分子结构式如下:英文名称:Capmatinib中文名称:卡马替尼本文主要对Capmatinib(卡马替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
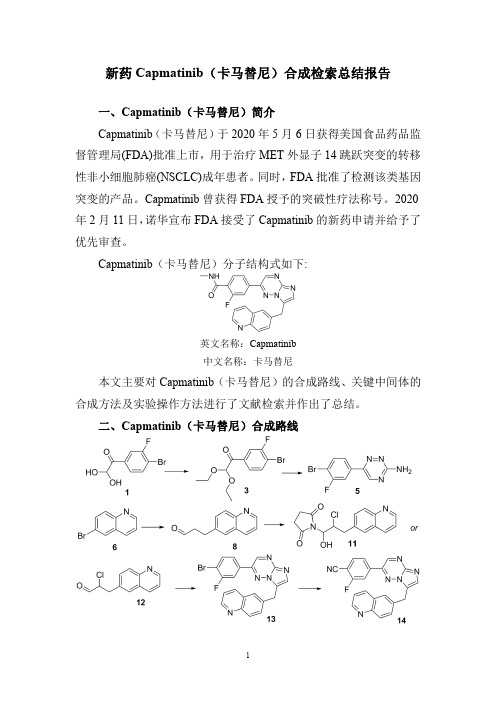
二、Capmatinib(卡马替尼)合成路线三、Capmatinib (卡马替尼)合成检索总结报告(一)Capmatinib (卡马替尼)中间体3的合成合成方法实验步骤参考文献操作方法一To a solution of 1-(4-bromo-3-fluorophenyl)-2,2-dihydroxy-ethanone 1(3.5g,0.014mol)in toluene (30mL)was added ethyl orthoformate 2(5.8mL,0.035mol)and p-toluene-sulfonic acid (100mg).The reaction was refluxed for 4h.After cooled to RT,the mixture was diluted with ethyl acetate,washed with saturated NaHCO 3solution,water,and brine,dried over MgSO 4,filtered and concentrated to give the product 3(4.0g,93%)which was used in the next step without further 2008/39457;(2008);(A1)English 操作方法二A 22L flask was charged with the hydrate of (4-bromo-3-fluorophenyl)-2-oxoacetaldehyde 1(1020g,4.41mol),toluene (7.5L),triethyl orthoformate 2(1633g,1.8L,11.04mol,2.5equiv),para-toluene sulfonic acid (33.5g,0.176mol,0.4equiv)at room temperature,and the resulting reaction mixture was heated to 110°C.and stirred at 110°C.for 6h.When HPLC showed that the reaction was complete,the reaction mixture was cooled down to room temperature before being poured into a 50L separation funnel along with ethyl acetate (7.5L)and the saturated aqueous sodium bicarbonate solution (NaHCO 3,3L).The mixture was stirred and the layers were separated.The aqueous layer was extracted with ethyl acetate (2L).The combined organic layers were washed with brine (4L),dried with sodium sulfate (Na 2SO 4),and concentrated under the reduced pressure to afford crude 1-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone 3(1240g,1345.7g theoretical,92.1%yield)which was used in the subsequent reaction without further 2009/291956;(2009);(A1)English(二)Capmatinib (卡马替尼)中间体5的合成合成方法实验步骤参考文献操作方法一A mixture of l-(4-bromo-3-fluorophenyl)-2,2-diethoxy-ethanone3(15.2g,50mmol),aminoguanidine bicarbonate4(10.2g,75mmol)and potassium hydroxide(6.6g,100mmol)in ethanol(200mL)and water(4mL)was refluxedovernight.The solvent was evaporated under reducedpressure and the residue was washed with acetonitrile andfiltered.The filtrate was concentrated under reducedpressure.The residue was dissolved in dichloromethane(100mL),washed with water,brine,and concentrated underreduced pressure.The residue was dissolved in ethanol(50mL).To the solution was added0.2N hydrochloric acid(50mL).The resultant mixture was heated to110o C for8h,andcooled with an ice-water bath.The precipitate that formedwas collected by filtration and washed with isopropanol togive the desired product5.(5.5g,41%)WO2008/64157;(2008);(A1)English操作方法二A22L flask was charged with1-(4-bromo-3-fluorophenyl)-2,2-diethoxyethanone3(1240g,4.07mol),ethanol(11L),water(1.4L),potassium hydroxide(KOH,910g,16.3mol,4.0equiv),and aminoguanidine bicarbonate4(1105g,8.13mol,2.0equiv)at room temperature.The resulting reactionmixture was then heated to75°C.for14h.When HPLCshowed the condensation reaction was deemed complete,thereaction mixture was cooled down to room temperaturebefore being filtered.The filtrate was then concentratedunder the reduced pressure to remove the most of thesolvents.The residual aqueous solution was extracted withethyl acetate(EtOAc,3×6L).The organic layers werecombined and concentrated under the reduced pressure togive a dark brown solid.This solid was dissolved in ethanol(4L)and the resulting solution was treated with a solution of0.2M aqueous hydrochloric acid solution(4L).Theresulting slurry was subsequently heated to50°C.for6hbefore being allowed to cool down to room temperature.Asolution of saturated aqueous sodium bicarbonate solution(NaHCO3,2L)was slowly added to the slurry and theresulting mixture was then concentrated under the reducedpressure to remove most of the solvents.The aqueousresidue was then treated with ethyl acetate(20L)to dissolveUS2009/291956;(2009);(A1)Englishthe solids.The two layers were separated andthe aqueous layer was extracted with ethyl acetate (2×2L).The combined organic layers were concentrated under the reduced pressure.The dark brown solids were treated with methyl tert-butyl ether (MTBE,4L)and the resulting slurry was heated to 30°C.and stirred at 30°C.for 30min.The mixture was filtered and the solids (green to orange in color)were collected (save the filtrate)and washed with methyl tert-butyl ether (MTBE,2L)to give the first crop of the crude desired product (5).The filtrate was evaporated under the reduced pressure,and the resulting dark brown solids were treated with methyl tert-butyl ether (MTBE,2L).The resulting slurry was heated to 30°C.and stirred at 30°C.for 30min.The mixture was filtered to give the second crop of the crude desired product (5)which was washed with MTBE (1L).The combined solids were dried in vacuum at 40-45°C.to afford 6-(4-bromo-3-fluorophenyl)-1,2,4-triazin -3-amine 5(585g,1095.1g theoretical,53.4%yield)which was used in the subsequent reaction without further purification.(三)Capmatinib (卡马替尼)中间体8的合成方法一合成方法实验步骤参考文献操作方法一Tris(dibenzylideneacetone)dipalladium (480mg,0.52mmol)and tri-tert-butyl-phosphonium tetrafluoroborate (300mg,1.0mmol)in a flask was evacuated and refilled with nitrogen (2times).1,4-dioxane (31mL)was added followed by consecutive addition of 6-bromoquinoline 6(7.2g,35mmol),2-propen-l-ol 7(4.7mL,69mmol)and N-cyclohexyl -N-methyl-cyclohexanamine (8.9mL,42mmol).The reaction vessel was evacuated and refilled with nitrogen (2times).The reaction mixture was stirred at 30o C for 24h.Diethyl ether (30mL)was added to the reaction mixture and then filtered and washed with diethyl ether.The organic extract was concentrated under reduced pressure.The residue was purified by flash chromatography eluting with ethyl acetate in hexanes (0-50%)to afford the desired product 8.(55%).WO2008/64157;(2008);(A1)English;US2011/212967;(2011);(A1)English.A 22L flask was charged with tris(dibenzylideneacetone)-dipalladium(0)(70.0g,0.076mol,0.015equiv),tri-tert-butylphosphonium tetrafluoroborate (44g,0.152mol,0.03。
新药Umbralisib(厄布利塞)合成检索总结报告
新药Umbralisib(厄布利塞)合成检索总结报告一、Umbralisib(厄布利塞)简介Umbralisib(厄布利塞)适应于治疗慢性淋巴细胞性白血病。
2020年5月5日,Therapeutics宣布了UNITY-CLL三期临床研究(评估了Umbralisib和Ublituximab联合治疗慢性淋巴细胞性白血病)达到阳性顶线结果。
Umbralisib(厄布利塞)分子结构式如下:英文名称:Umbralisib中文名称:厄布利塞本文主要对Umbralisib(厄布利塞)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Umbralisib(厄布利塞)合成路线三、Umbralisib(厄布利塞)合成检索总结报告(一)Umbralisib (厄布利塞)中间体3的合成合成方法实验步骤参考文献操作方法一Potassium acetate(10.52g,107.2mmol)and bis(pinacolato)diboron 2(15g,58.96mmol)were added to a solution of intermediate 1(10.52g,107.2mmol)in dioxane (125ml),and the solution was degassed for 30min.[1,1'-Bis(diphenylphosphino)ferrocene]dichloro palladium(II).CH 2Cl 2(4.4g,5.36mmol)was added under nitrogen atmosphere and heated to 80°C.After 12h,the reaction mixture was filtered through celite and concentrated.The crude product was purified by column chromatography with ethyl acetate :petroleum ether to afford the title compound 3as a yellow oil (13.9g,99%)which was used without purification in the next 2012/289496;(2012);(A1)English;WO2014/6572;(2014);(A1)English;WO2012/151525;(2012);(A1)English;US2015/290317;(2015);(A1)English(二)Umbralisib (厄布利塞)中间体5的合成合成方法实验步骤参考文献操作方法一To a solution of 3-iodo-1H-pyrazolo[3,4-d]pyrimidin-4-amine 4(11.0g,42.14mmol)in DMF 110ml),ethanol (55ml)and water (55ml),intermediate 3(23.4g,84.28mmol)and sodium carbonate (13.3g,126.42mmol)were added and degassed for 30min Tetrakis(triphenylphosphine)palladium (0)(2.4g,2.10mmol)was added under nitrogen atmosphere and heated to 80°C.After 12h,the reaction mixture was filtered though celite,concentrated and extracted with ethyl acetate.The organic layer was dried over sodium sulphate and concentrated under reduced pressure.The crude product was triturated with diethyl ether,filtered and dried under vacuum to afford the title compound as light brown solid 5(3.2g,26%yield)which is used as such for the next 2012/289496;(2012);(A1)English;WO2014/6572;(2014);(A1)English;WO2012/151525;(2012);(A1)English;US2015/290317;(2015);(A1)English(三)Umbralisib (厄布利塞)中间体8的合成。
药物帕比司他(panobinostat)合成检索总结报告
药物帕比司他(panobinostat)合成检索总结报告
一、帕比司他(panobinostat)简介
帕比司他(panobinostat)联合硼替佐米和地塞米松用于治疗既往接受至少两种治疗方案(包括硼替佐米和一种免疫调节药物)的多发性骨髓瘤(MM)患者。
帕比司他(panobinostat)不良反应有:腹泻,疲劳、恶心、外周性水肿、食欲下降、发热、呕吐、低磷酸盐血症,低钾血症、低钠血症等等。
帕比司他(panobinostat)分子结构式如下:
英文名称:panobinostat 英文名称:panobinostat Lactate
中文名称:帕比司他中文名称:帕比司他乳酸盐本文主要对帕比司他(panobinostat)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、帕比司他(panobinostat)合成路线
三、帕比司他(panobinostat)合成检索总结报告
(一) 帕比司他(panobinostat)中间体3的合成。
药物Cobimetinib(考比替尼)合成检索总结报告
药物Cobimetinib(考比替尼)合成检索总结报告
一、Cobimetinib(考比替尼)简介
Cobimetinib(考比替尼)与维罗非尼(Vemurafenib)联用用于治疗BRAF V600E或V600K变异的不可切除或转移性黑素瘤癌患者。
Cobimetinib(考比替尼)有不良反应有:腹泻,光敏反应,恶心,发热和呕吐。
Cobimetinib(考比替尼)分子结构式如下:
英文名称:Cobimetinib
中文名称:考比替尼
本文主要对Cobimetinib(考比替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Cobimetinib(考比替尼)合成路线
三、Cobimetinib(考比替尼)合成检索总结报告(一) Cobimetinib(考比替尼)中间体2的合成
(二) Cobimetinib(考比替尼)中间体3的合成
(三) Cobimetinib(考比替尼)中间体5的合成
(四) Cobimetinib(考比替尼)中间体6的合成。
新药Cabozantinib(卡博替尼)合成检索总结报告
新药Cabozantinib(卡博替尼)合成检索总结报告一、Cabozantinib(卡博替尼)简介Cabozantinib(卡博替尼)是一个多靶点小分子酪氨酸激酶抑制剂。
Cabozantinib(卡博替尼)的靶点包括MET、ROS1、RET、AXL、NTRK、KIT等九大靶点。
目前,Cabozantinib(卡博替尼)已经在甲状腺髓样癌、肾癌、非小细胞肺癌、肝癌、软组织肉瘤、前列腺癌、乳腺癌、卵巢癌、肠癌等多种实体瘤中,证实了较好的治疗效果,对于骨转移的控制效果尤其突出。
Cabozantinib(卡博替尼)分子结构式如下:英文名称:Cabozantinib中文名称:卡博替尼本文主要对Cabozantinib(卡博替尼)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Cabozantinib(卡博替尼)合成路线三、Cabozantinib(卡博替尼)合成检索总结报告(一)Cabozantinib (卡博替尼)中间体3的合成合成方法实验步骤参考文献操作方法一The intermediate 1(45.0g,0.2mol)was dissolved in chlorobenzene (450mL,10v/w),4-nitrophenol 2(70.1g,0.50mol)and N,N-diisopropylethylamine (51.7g,0.4mol)were sequentially added.After stirring at 140o C for 14h,the reaction mixture was cooled to r.t.,The precipitates were collected by filtration and washed with appropriatepetroleum ether to afford compound 3as a yellow solid in 88.2%yield.Bioorganic and Medicinal ChemistryLetters ;vol.29;nb.19;(2019);Art.No:126630.操作方法二To chloroquinoline 1(0.24g,1.1mmol)in diphenyl ether (20mL)was added 4-nitrophenol 2(0.30g,2.2mmol)and the resulting mixture was heated to 170°C.for 24h.The mixture was partitioned between ethyl acetate and 1M NaOH(aq).The organic phase was collected,washed with water and brine,dried (MgSO 4),filtered and concentrated.The residue was purified by flash column chromatography (90%ethyl acetate/hexanes-ethyl acetate)to afford 3(0.25g,69%)as a colorless solid.US2008/4273(2008);(A1)English操作方法三4-Nitrophenol 2(12.5g,89.6mmol)was added into a suspension of 4-chloro-6,7-dimethoxyquinoline 1(10.0g,44.8mmol)in PhCl (80mL).The resulting mixture was stirred at reflux for 16h.The solvent was evaporated under reduced pressure,and the residue was dissolved in CH 2Cl 2(150mL)The solution was washed by 10%NaOH aqueous solution (3×30mL),water (30mL)and dried (MgSO 4),and evaporated to obtain the title compound 3as a yellow solid (9.6g,65.7%)without further purification.Bioorganic and MedicinalChemistry ;vol.27;nb.17;(2019);p.3825–3835.操作方法四4-chloro-6,7-dimethoxyquinoline 1(671mg,3.0mmol)and 4-nitrophenol 2(500mg,3.6mmol)were placed in 7mL of chlorobenzene.Heat slowly to 140°C and continue to react at this temperature for 20h.Then the heating was stopped,cooled to room temperature,the solvent was evaporated under reduced pressure,and the residue was dissolved in dichloromethane.Then washed successively with saturated potassium carbonate solution,washed with water,dried over anhydrous sodium sulfate,and concentrated under reduced pressure,purified by silica gel column chromatography (PE /CN109988110;(2019);(A)Chinese.EA =3:1),to give apale yellow solid (3)620mg,63%yield.操作方法五To a suspension of 4-chloro-6,7-dimethoxyquinoline 1(40g,0.18mol)and 4-nitrophenol 2(26.2g,0.19mol)in toluene (60mL)was added DIPEA (27.8g,0.22mol).The reaction was heated to 115°C for 24hours and then concentrated in vacuo.The residue was washed with EtOH (40mL)to give the title compound 3as a pale yellow solid (28g,47.8%).WO2013/180949;(2013);(A1)English操作方法六A reactor was sequentially charged with 4-chloro-6,7-dimethoxy-quinoline 1(8.0kg),4-nitrophenol 2(7.0kg),4-dimethylaminopyridine (0.9kg),and 2,6-lutidine (40.0kg).The reactor contents were heated to approximately147°C.When the reaction was complete (less than 5percent starting material remaining as determined by in process HPLC analysis,approximately 20hours),the reactor contents were allowed to cool to approximately 25°C.Methanol (26.0kg)was added,followed by potassiumcarbonate (3.0kg)dissolved in water (50.0kg).The reactor contents were stirred for approximately 2hours.Theresulting solid precipitate was filtered,washed with water (67.0kg),and dried at 25°C for approximately 12hours to afford the title compound 3(4.0kg).WO2015/164869;(2015);(A1)English;EP2017/2758057(2017);(B1)English操作方法七523g of p-nitrophenol 2(3.76mol)was dissolved in 600ml (6.45mol)of N,N-dimethylacetamide,(4.3mol)of potassium t-butoxide and 800g (3.58mol)of4-chloro-6,7-dimethoxyquinoline 1and 1.5L (16.1mol)of N,N-Dimethylacetamide solution,and the reaction solution was heated to 100°C to 120°C and reacted for 2hours.The reaction solution was cooled to room temperature,poured into 3.5L ice water,stirred for 1to 2hours and then filtered.The filter cake was washed twice with 2L of water and then dried in vacuo at 35°C,To give 6,7-dimethoxy-4-(4-nitro-phenoxy)quinoline 3as a pale yellowish white powder 918.2g,the molar yield was 78.6%.CN103664778;(2017);(B)Chinese(二)Cabozantinib (卡博替尼)中间体4的合成合成方法实验步骤参考文献To 3(0.25g,0.77mmol)in 1:1MeOH/THF (50mL)was added Zn dust (0.55g,8.4mmol)and ammonium chloride操作方法一(0.085g,1.6mmol)in water(5mL).The resulting mixturewas heated to reflux for2h,then filtered through celite and concentrated.The residue was dissolved in dichloromethane,washed with water,brine,dried(MgSO4),filtered andconcentrated to provide crude4(0.25g,>100%)which wasused without further purification.US2008/4273(2008);(A1)English操作方法二500ml of methanol was added to the autoclave,and100g of intermediate3and25g of Raney nickel were added.Raisethe temperature at30°C for10hours;Press filtration,concentration,crystallization,filtration,and drying gave86gof Intermediate4in a yield of95%.CN108264482;(2018);(A)ChiNese;CN110240563;(2019);(A).操作方法三Conc hydrochloric acid(11mL,0.2v/w)and iron powder(56.7g,1.0mol)were sequentially added into90%ethanol(550mL,10v/w)under stirring for10min.Then thereaction mixture was heated to60o C,the intermediate3(55.0g,0.17mol)was added.After refluxing for1h,activatedcarbon(1.5g)was added and then reflux for30min.Themixture was filtered while hot,the filtrate was cooled toroom temperature,adjusted to pH12with10N NaOH,poured into water and stirred for2h.The precipitates werecollected by filtration and washed with water until the filtratewas nearly neutral to obtain compound4as a pale yellowsolid in90.7%yield.m.p.:196.4-197.2o C.Bioorganic andMedicinalChemistryLetters;vol.29;nb.19;(2019);Art.No:126630.操作方法四A mixture of3(9.6g,29.4mmol),Fe(8.2g,0.15mol)andAcOH(0.5mL)in90%EtOH(100mL)was refluxed withvigorous agitation for4h.The hot solution was filteredthrough celite and the filter cake was washed with hot EtOH(20mL).The combined filtrate was concentrated underreduced pressure to afford a dark brown solid,which wasrecrystallized from EtOH to afford4as yellow solid(7.2g,82.9%).Bioorganic andMedicinalChemistry;vol.27;nb.17;(2019);p.3825-3835操作方法五6,7-dimethoxy-4-(4-nitrophenyloxy)quinoline3(620mg,1.9mmol)was dissolved in ethanol(40mL).After beingdissolved by stirring,tin(II)chloride dihydrate(1.25g,4.9mmol)was added in portions.After the addition wascompleted,slowly increase to70°C for6h.After thereaction was completed,the reaction solution was cooled toroom temperature and diluted with a1N NaOH(10mL)aqueous solution.It was extracted with ethyl acetate(3×15mL),and the organic layer was combined and washedsequentially with1N aqueous NaOH,water and saturatedaqueous sodium chloride.Dried over anhydrous sodiumsulfate,filtered,and concentrated under reduced pressure toyield a yellow solid355mg,60%yield.CN109988110;(2019);(A)Chinese。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
药物Copanlisib(库潘尼西)合成检索总结报告一、Copanlisib(库潘尼西)简介Copanlisib(库潘尼西)于2017年9月在美国上市,主要用于治疗病情复发的滤泡性淋巴瘤成人患者。
Copanlisib(库潘尼西)常见的不良反应有高血糖、腹泻、体力下降、高血压、白细胞减少、恶心、下呼吸道感染、血小板减少症。
Copanlisib(库潘尼西)分子结构式如下:CAS:1032568-63-0CAS:1402152-13-9英文名称:Copanlisib英文名称:Copanlisib Dihydrochloride中文名称:库潘尼西中文名称:库潘尼西盐酸盐本文主要对Copanlisib(库潘尼西)的合成路线、关键中间体的合成方法及实验操作方法进行了文献检索并作出了总结。
二、Copanlisib(库潘尼西)合成路线三、Copanlisib (库潘尼西)合成检索总结报告(一)Copanlisib (库潘尼西)中间体2的合成合成方法实验步骤参考文献操作方法一Small 12g (0.06mole)portions of compound 1are slowly added to 50ml of a fuming nitric acid solution cooled in an ice bath to 0°C.The reaction mixture is stirred for 1hour at a temperature of less than 5°C.The reaction mixture is then poured into the ice water and stirred again for 1hour.The product 2is isolated by vacuum filtration and cyclohexane washing (yield 80%-11.5g).US2015/266845;(2015);(A1)English 操作方法二To fuming HNO 3(600mL)is added 4-formyl-2-methoxy-phenyl acetate 1(10.0g,51.2mmol)in portions at -10°C,and the mixture is stirred for 45min.The acidic solution is slowly poured into ice-water (1L)and the precipitated product is collected by filtration.The precipitate is washed several times with ice water (250mL)and dried.The crude product is recrystallized from EA/PE (3:7)to give the desired product 2(8.00g,yield 75%)as yellow needles.WO2014/149793;(2014);(A1)English 操作方法三To a solution of 4-formyl-2-methoxyphenyl acetate 1(30.0g,154.5mmol,1.0eq)in DCM (300.0mL)was added fuming nitric acid (154.5mmol,1.0eq)at -30°C.The mixture was stirred at 20°C for 15hrs.The reaction mixture was quenched by addition water 100mL at 0°C,and then diluted with DCM 50mL and extracted with DCM (50mL×3).The combined organic layers were washed with brine (100mL×2),dried over Na 2SO 4,filtered and concentrated under reduced pressure to give a residue.The residue was purified by column chromatography (SiO 2,Petroleum ether/Ethyl acetate=30/1to 10:1)to give 4-formyl-2-meth-oxy-3-nitrophenyl acetate 2(23.0g,96.2mmol,62.2%yield)as a white solid.WO2017/151489;(2017);(A1)English操作方法四Fuming nitric acid (2200mL)undernitrogen was cooled to 0o C at which time vanillin acetate 1(528g,2.7mol)was added portionwise,keeping the internal temperature below 10o C.After 2h the resulting mixture was poured over ice with stirring.The slurry was filtered and the resulting solids were washed with water (3×100mL)and air-dried.After 2days the solids were heated in DCM (3000mL)until complete dissolution.The solution was allowed to cool to room temperature while hexanes (3000mL)was added dropwise.The solids were filtered,washed with hexanes (500mL)and air dried to give the desired product 2(269g,41%).WO2008/70150;(2008);(A1)English;WO2012/62748;(2012);(A1)English;WO2012/62743;(2012);(A1)English;WO2012/62745;(2012);(A1)English(二)Copanlisib (库潘尼西)中间体3的合成合成方法实验步骤参考文献操作方法一24g acetyl-2-nitrovanilline 2were suspended in 300ml water at 10-30o C.The mixture was heated to 40-45o C and 40g NaOH 30%(300mmol,3equiv.)were added in 10-20min.Stirring was continued for 30-45min.The reaction was cooled to 25-30o C and the pH was set to 2.0-2.5by the addition of 10%sulfuric acid.Stirring was continued for 15min at 20-25o C and the product was filtered off and washed with water (2×100ml).The product was dried at 45-50o C under reduced pressure and 18.7g 2-nitrovanilline 3were obtained a white to yellow solid in 95%yield.WO2016/71380;(2016);(A1)English 操作方法二A mixture of 4-formyl-2-methoxy-3-nitrophenyl acetate (2)438g (1.8mol)and potassium carbonate (506g,3.7mol)in MeOH (4000mL)was stirred at room temperature for 16h.The reaction mixture was concentrated under reduced pressure to afford a viscous oil.This was dissolved in water,acidified using a solution of HCl (2N)and extracted with EtOAc.The organic layer was washed with brine,dried (MgSO 4)and filtered.The solvent was concentrated under reduced pressure to 1/3volume and the resulting solids were filtered and air-dried to give the title compound 3(317g,88%).WO2008/70150;(2008);(A1)English;WO2012/62748;(2012);(A1)English;WO2012/62743;(2012);(A1)English;WO2012/62745;(2012);(A1)10g of compound 2(0.042mole)are added to a solution of 40ml of sodium hydroxide 33%(m/m).The reaction mixture is heated at reflux for 10minutes.The reactionUS2015/266845;。