呼吸系统肿瘤用药 塞瑞替尼 ceritinib
英-[药物报告]色瑞替尼
![英-[药物报告]色瑞替尼](https://img.taocdn.com/s3/m/6a60568adaef5ef7ba0d3ca9.png)
Ceritinib(Zykadia ®)Research code: LDK-3781 General Information● Ceritinib is a kinase inhibitor, which was first approved in2014 by US FDA.● Ceritinib was discovered and marketed by Novartis. ● Ceritinib inhibited autophosphorylation of ALK, ALK-me-diated phosphorylation of the downstream signaling protein STAT3, and proliferation of ALK-dependent cancer cells.● Indicated for the treatment of patients with anaplastic lym-phoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intol-erant to crizotinib.● Available as capsules with each containing 150 mg ofceritinib and recommended dose is 750 mg once daily until disease progression or unacceptable toxicity.Key Approvals around the World *First approvaldate 04/29/2014 06/05/2015 NDA NO. 205755 EMEA/H/C/003819Brand name Zykadia ® Zykadia ® Indication NSCLC NSCLCAuthorisation holderNovartisNovartis Europharm Ltd*Till Dec 2015, it has not been approved by PMDA (Japan) and CFDA (China).Worldwide SalesActive IngredientMolecular formula : C 28H 36N 5O 3ClS Molecular weight : 558.14 CAS No.: 1032900-25-6Chemical name : 5-Chloro-N 4-[2-[(1methylethyl)sul-fonyl]phenyl]-N 2-[5-methyl-2-(1-methylethoxy)-4-(4-pi-peridinyl)phenyl]-2,4-pyrimidinediamine Parameters of Lipinski's “Rule of 5”5583871144.70 ± 0.95aCalculated by ACD/Labs software V11.02.Drug Product *Dosage route : Oral Strength : 150 mg Dosage form : CapsuleInactive ingredient : Colloidal anhydrous silica, L-hydroxy-propylcellulose, magnesium stearate, microcrystalline cel-lulose, and sodium starch glycolate, gelatin, indiogotine, and titanium dioxide Recommended dose :The recommended starting dose is 750 mg once daily with-out food.Discontinue ceritinib for patients unable to tolerate 300 mg daily.If a dose of ceritinib is missed, make up that dose unless the next dose is due within 12 h.*Sourced from the FDA drug label information.2 ▐2014 Worldwide NCEsKey PatentsPatents Summary•Ceritinib’s compound patent application was filed as PCT application by IRM in 2007.•The compound patent will be expired in 2027 originally, which has been granted in Japan, Europe, the United States successively.•Zykadia® (Ceritinib) has got five years NCE market exclusivity protection and seven years ODE after it was approved by FDA in Apr. 29, 2014 initially.Patents ListCeritinib ▐ 32 ChemistryRoute 1: Original Discovery RouteSynthetic Route: The overall synthetic route of ceritinib was enabled via a modified Buchwald-Hartwig coupling of two advanced intermediates aryl amino-piperidine 6 and arylsulfonyl chloro-pyrimidine 7. Aryl amino-piperidine 6 was synthesized in five stepsfrom 2-chloro-4-fluorotoluene 1. Nitration of 1 using KNO3/H2SO4 yielded the corresponding nitro derivative 2 in a 71% yield. Condensation of compound 2 with iso propanol in the presence of Cs2CO3 provided the 2-chloro-4-iso propoxy-5-nitrotol-uene 3 in excellent yield. Suzuki coupling of compound 3 with 4-pyridineboronic acid 4 provided compound 5 in 73% yield. Concomitant reduction of pyridine and nitro group using PtO2/H2 afforded the corresponding piperidine-aniline intermediate, which was subsequently protected with Boc, generating compound 6 in 60% over two steps. Buchwald-Hartwig coupling of 6 with arylsulfonyl chloro-pyrimidine 7 provided the precursor of ceritinib. Boc deprotection with TFA, concentration, and subse-quent precipitation with 1 N HCl yielded ceritinib as the HCl salt in 35.0% yield from 6 and the overall yield of 10.3%.[1, 2] Synthesis of the required arylsulfonyl chloro-pyrimidine coupling precursor 7 began with 2-(isopropylsulfonyl)benzenamine 8 and 2,4,5-trichloropyrimidine 9 under basic condition (NaH, DMF, DMSO)in 60% yield.4 ▐2014 Worldwide NCEsRoute 2:Synthetic Route: In this strategy, ceritinib was prepared from reduction of 4-(5-isopropoxy-2-methyl-4-nitrophenyl)pyridine 1 under Pd/C H2 condition afforded corresponding phenylamine 2 in 92% yield. Then Sandmeyer reaction was carried out in the presence of NaNO2/CuBr/HBr/H2O to give 4-(4-bromo-5-isopropoxy-2-methylphenyl)piperidine 3 in 83% yield. Buckward-Hartwig coupling of 3 with 5-chloro-4-nitropyrimidin-2-amine 4 converted to desired compound 5 in 78% yield, which was hy-drogenated and coupled with 1-bromo-2-(isopropylsulfonyl)benzene 7 to obtain the target ceritinib in 74% yield over two steps. The overall yield was 44%.[3]3 PharmacologySummaryMechanism of Action●Ceritinib is a kinase inhibitor, which inhibited ALK, insulin-likegrowth factor 1 receptor (IGF-1R), insulin receptor (InsR), andROS1.●Ceritinib was approximately 50-fold more specific for ALK(IC50 = 0.15 nM) than InsR (IC50 = 7 nM) and IGF-1R (IC50 = 8nM), other members of theinsulin receptor superfamily.●Ceritinib inhibited autophosphorylation of ALK, ALK-mediatedphosphorylation of the downstream signaling protein STAT3,and proliferation of ALK-dependent cancer cells.●The off-target activity of ceritinib was evaluated for 42 of thetarget receptors (MC4, IC50 = 0.6 nM), 84 of the broad spectrumscreen receptors (transporter monoamine, inhibition = 97%), and10 of the GPCRs (dopamine 2 receptor, IC50 = 4000 nM).Ceritinib ▐ 5 In Vitro Efficacy●Anti-proliferative activity of ceritinib in tumor cell lines:Cell lines expressed with ALK fusion protein: IC50 = 11-56 nM.Cell lines expressed with EML4-ALK-mutation: IC50 = 37.6-250 nM.Ba/F3 cells expressed with others fusion proteins: IC50 = 180-400 nM.●Phosphorylation of ceritinib in Karpas299 cells:ALK protein: IC50 = 46 nM.STAT3 protein: IC50 = 150 nM.In Vivo Efficacy●H2228 cells xenograft models:In SCID mice: Significance at doses ≥6.25 mg/kg. Complete tumor regression with 25 mg/kg ceritinib after 14 days.In nude rats: Significance at doses ≥10 mg/kg.●Karpas299 cells xenograft models:In SCID mice: Significance at doses ≥12.5 mg/kg. Complete tumor regression with 25 mg/kg ceritinib after 14 days.In nude rats: Significance at doses ≥12.5 mg/kg. Complete tumor regression with 25 mg/kg ceritinib after 14 days.●Crizotinib-resistant H2228 cells xenograft model carrying the ALK-mutation in SCID mice:Non-ALK-mutation: Significant at doses ≥50 mg/kg.I1171T ALK-mutation: Significant at doses ≥25 mg/kg.C1156Y ALK-mutation: Significant at doses ≥50 mg/kg.Mechanism of ActionTable 1In Vitro Effects of Ceritinib and Crizotinib on Human Protein Kinases[4]EPK CE ALK (1066-1459) Y a0.15 3 EPK CE AXL Y 180 13EPK CE IGF-1R (980-1369) Y 8 400 EPK CE RET (658-1072) Y 400 2200 EPK CE INSR (871-1343) Y 7 290 EPK ROCK2 S/T 450 2500EPK AURORA_A S/T b110 600 EPK CE FGFR3(411-K650E-806) Y 430 1700EPK cABLT315 Y 130 6The applicant evaluated the selectivity of ceritinib and crizotinib by testing its in vitro activity against 36 recombinant human protein kinases using the Caliper mobility shift assay, table 1 showed ceritinib most selective for ALK (IC50 <0.5 μM). a Y: Tyrosine-specific protein kinases. b S/T: Serine/Threonine-specific protein kinases.[4]H2 receptor 170 NAmonoamineRabbit 10 μM 97 331Opk receptor 570 NA K channel[Ka] Rat 10 μM 99 682MC3 receptor 670 NA Somatostatin sst1 Human 10 μM 88 2420MC4 receptor 600 35% at 10 μM Somatostatin sst2 Human 10 μM 76 2250Ad3 receptor 730 8300 Somatostatin sst3 Human 10 μM 93 5120NK1 receptor 990 79% at 30 μM Somatostatin sst4 Human 10 μM 90 1880D2 receptor 1200 NA 5-HT5A Human 10 μM 71 5000The off-target activity of ceritinib was evaluated using an in vitro safety pharmacology profiling panel including 139 G protein-coupled receptors (GPCRs), ion channels, nuclear receptors, transporters, and enzymes that have been previously linked to potential side effects. At least eight concentrations of ceritinib were tested. The agonist and antagonist activities of ceritinib were evaluated for 10 GPCRs using a safety pharmacology screen. A fluorometric imaging plate reader (FLIPR) assay was conducted with a four-fold, eight-point serial dilution of ceritinib up to 10 μM to a ssess agonist and antagonist activity. Ceritinib showed weak agonist activity on the dopamine D2 receptor (EC50 = 6 μM, IC50 = 4 μM), but did not exhibit significant activity against the other GPCRs tested. a Ceritinib interacted with 42 of the target receptors, table 2 showed the target receptors IC50 <1500 nM. b A safety pharmacology broad spectrum screen with 84 receptors at ceritinib was tested at 10 μM in duplicate, table 2 showed the broad spectrum receptors inhibited >70%.6 ▐2014 Worldwide NCEsIn Vitro EfficacyTable 3 In Vitro Anti-proliferative and Phosphorylation of Ceritinib[4]Anti-proliferative activityBa/F3Murinepro-B lymphoma cell withALK fusion kinaseNPM-ALK 26-35NATEL-ALK-Q1 56EML4-ALK 27 Ba/F3Murinepro-B lymphoma cell withALK-mutationEML4-ALK I1171T 37.6 440EML4-ALK L1196M 69 1460EML4-ALK G1202R 94 1370EML4-ALK C1156 160 340EML4-ALK S1206A 160 270EML4-ALK G1269S 250 2140 Ba/F3Murinepro-B lymphoma cell withtyrosine kinaseTEL-ROS 180NATEL-IGF-1R 220TEL-InsR 400Karpas299 Human ALCL NPM-ALK 45 (n = 8)NA NCI-H2228 Human NSCLC EML4-ALK 11 (n = 5) NB-1 Human neuroblastoma cell ALK 24 (n = 3)Endogenous phosphorylation Karpas299 Human ALCLALK protein 46NASTAT3 protein 150The cell viability following 48 h of treatment with control compounds or ceritinib serially diluted with DMSO was determined using the Bright-Glo™ luciferase biolumi-nescent assay and cell proliferation was measured using the Brite Lite™ luciferase assay. ALCL: Anaplastic large-cell lymphoma. NSCLC: Non-small cell lung cancer. NA: Not available.In Vivo EfficacyTable 4 In Vivo Efficacy of Ceritinib in Different Xenograft Models[4]H2228 HumanNSCLCSCIDmouse(female)Wild type3.125 QD × 14416.25Complete tumor regressionwas observed even at adose of 25 mg/kg/day af-ter 14 days.6.25 QD × 143612.5 QD × 14-6425 QD × 14-100RNU nuderat(female)Wild type5 QD × 146710Complete tumor regressionwas observed even at adose of 25 mg/kg/day af-ter 14 days.10 QD × 14 225 QD × 14-100Ceritinib ▐ 7ContinuedC1156Y ALK-mu-tation25 QD × 12 61.8 50 Complete tumor regres-sion was observed even at a dose of 100 mg/kg/day.50 QD × 12 11.7 100 QD × 12 -100 Karpas299Human ALCLSCID mouse (female)Wild type6.25QD ×14 62 12.5 Complete tumor regres-sion was observed at 25 mg/kg after 14 mg/kg/day.12.5 QD × 14 18 25 QD × 14 -93 RNU nuderat (female)Wild type6.25QD × 14 74 12.5Significant tumor regres-sion was observed at 25 mg/kg/day after 14 days.12.5QD × 143025 QD × 14 -33 50QD × 14-66Study: H2228 NSCLC xenograft mice model Animal: SCID beige mice (female)Model: 5 × 106 NCI-H2228 cells were implanted subcutaneously into the right hind flank of one SCID mouse.Administration: Treated daily, p.o., ceritinib: 3.125, 6.25, 12.5 and 25 mg/kg/day, ve-hicle control: NAStarting treatment: Mice bearing established tumors (~85 mm 3) Test: Tumor volumes, 3 times a weekResult: Treatment with 25 mg/kg ceritinib resulted in complete tumor regression after 14 days. ** P <0.05.Figure A Effect of Ceritinib on Human H2228 NSCLC Xenografts in SCID Mice Model [4]8 ▐2014 Worldwide NCEsStudy: Crizotinib-resistant H2228 cells xenograft with ALK I1171T mutations micemodelAnimal: SCID beige mice (female)Model: Crizotinib-resistant H2228 NSCLC with ALK I1171T mutations were im-planted subcutaneously into one SCID mouse.Administration: Treated daily, p.o., ceritinib: 12.5, 25, or 50 mg/kg/day, crizotinib100 mg/kg/day, vehicle control: NAStarting treatment: Mice bearing established tumors (~130 mm3)Test: Tumor volumes, 3 times a weekResult: Treatment with 50 mg/kg ceritinib resulted in complete tumor regression after14 days of treatment. (**P≤0.05; ****P≤0.0001)Figure B Effect of Ceritinib on Crizotinib-resistant Human H2228 NSCLC Xenograft with ALK I1171TMutations in SCID Mice Model[4]Study: Karpas299 ACLC xenograft mice modelAnimal: SCID beige mice (female)Model: 5 × 106 Karpas299 cells were implanted subcutaneously into the right hindflank of one SCID mouse.Administration: Treated daily, p.o., ceritinib: 6.25, 12.5 and 25 mg/kg/day, vehiclecontrol: NAStarting treatment: Mice bearing established tumors (~74 mm3)Test: Tumor volumes, 3 times a weekResult: Treatment with 25 mg/kg ceritinib resulted in c significant tumor regressionafter 14 days of treatmentFigure C Effect of Ceritinib on Human Karpas299 ALCL Xenografts in SCID Mice Model[4]4 ADME & Drug-Drug InteractionSummaryAbsorption of Ceritinib●Exhibited a non-linear pharmacokinetics in humans after oral administration. The increase in AUC appeared to be greaterthan dose-proportional in the dose range of 50 to 750 mg ceritinib.●Had a moderate bioavailability in rats (48.3%), but high in mice (54.6%) and dogs (58%).●Was observed slowly (T max = 3.98-15 h) in humans, mice (7 h), rats (12 h) and monkeys (13-18.3 h).●Showed a half-life of 19.4-40.6 h in humans, much longer than those in rats (13.2 h) and monkeys (12.1-16 h), after oral ad-ministration.●Had a moderate clearance in mice (26.6 mL/min/kg), rats (1.49 L/h/kg), but low to moderate in monkeys (0.366-0.78 L/h/kg),after intravenous administration. The Cl/F in humans was 44.5-126 L/h after oral administration.●Exhibited an extensive distribution in mice, rats and dogs, with the apparent volume of distribution at 9.7, 19.9 and 6.53-13.5L/kg, after intravenous administration. The V z/F in humans was 1880-6230 L after oral administration.●Ceritinib was classified as a low passive permeability compound.Distribution of Ceritinib●Exhibited high plasma protein binding in rats (97.9-98.4%), dogs (98.3-98.4%), monkeys (94.4-97.4%) and humans (96.7-98.8%). Note that ceritinib was mainly bound to HSA.●The binding to RBC was 56.9-58.6% in humans, indicating the drug was distributed more to blood cells than to plasma.●In Long Evens rats after oral and intravenous administration:Ceritinib ▐ 9 The drug was widely distributed into most tissues including central nervous system although with a lower degree com-pared to other tissues.Relatively higher concentration levels were observed in intestinal wall, uveal tract, pituitary gland, bile, adrenal cortex, harderian gland, liver, spleen, lymph node, lungs, kidneys, thyroid, bone marrow, adrenal medulla, pancreas, thymus and salivary gland. The concentration in tissues was generally higher than blood concentration.The radioactivity concentration in the uveal tract was 176 times of that in blood, suggesting a high affinity for melanin-rich tissues.Ceritinib-derived radioactivity was retained in several other tissues (including testis, epididymis, and skin) and the elimi-nation was not yet complete by 168 hours post-dose.Metabolism of Ceritinib●Could slightly metabolized in rat, monkey and human hepatocytes.●CYP3A was the major metabolizing enzymes, with CYP2C19, 1A2, 2C8, 2D6 and 2C9 involved in the metabolism ofceritinib.●The metabolism of ceritinib included mono-oxygenation, O-dealkylation, S-dealkylation, and N-formylation of ceritinib, andsecondary di-oxygenation, glucuronidation, sulfation and dehydrogenation.●Overall, the parent drug was the most abundant component in plasma in humans. Eleven metabolites were found in theplasma of humans, each at levels ≤ 2.3% of the total drug-related AUC.●Five of these eleven metabolites were not detected in the plasma of either the rat or monkey. The remaining three uniquehuman metabolites detected at low levels in plasma included M46.6 (1.7%), M48.8 (1.7%), and M52.0 (2%).Excretion of Ceritinib●Was predominantly eliminated in feces in rats, monkeys and humans, with the parent drug as the significant component in rat,monkey and human feces.●In cannulated rats, excretion was mainly into the bile 65.4% after oral administration and 24.3% after intravenous administra-tion.Drug-Drug Interaction●Ceritinib was a strong inhibitor of CYP3A4/5 (IC50= 0.2 μM), and moderate of CYP2A6 (IC50= 5 μM), CYP2B6 (IC50 = 2μM), CYP2C8 (IC50= 25 μM), CYP2C9 (IC50= 2 μM), CYP2C19 (IC50= 70 μM), CYP2D6 (IC50 = 20 μM) and CYP2E1 (IC50= 30 μM).●Ceritinib had no induction for CYP1A2, CYP2B6 or CYP3A4 mRNA/activities, but had induction for CYP2C9 in vitro at theconcentration of 0.25-2.5 μM ceritinib.●Ceritinib was a substrate of P-gp and BCRP in vitro, but not inhibitors of them.●Ceritinib was not a substrate of OCT1, OAT2, OATP1B1 or OATP2B1.●Ceritinib inhibited OATP1B1 or OATP1B3, OAT1 and OCT2, with the IC50 of >5 μM in vitro, but did not inhibit OAT3 orOCT1.AbsorptionTable 5 In Vivo Pharmacokinetic Parameters of Ceritinib in Mice, Rats and Monkeys after a Single Dose Administration[4]Balb/c mouse (male) i.v. 5 -nM nM·h nM·h a6.2 ± 0.5mL/min/kg9.7 ± 0.6 - p.o. 20 7.0 ± 0695 ± 31nM12296 ± 981nM·h10334 ± 963nM·h a- - - 54.6 ± 4.4HanWistar rat (male) i.v. 10 0.083 ± 0 975 ± 139 6950 ± 1470 6890 ± 1510 9.7 ± 1.2 1.49 ± 0.342 19.9 ± 0.49 - p.o. 25 12.0 363 8390 8330 13.2 NA NA 48.3Cynomolgus monkey (male) i.v. 5 0.083 1410 6530 6450 29 0.78 13.5 -i.v. 10 0.083 3190 27800 27400 14.5 0.366 6.53 -p.o. 30 18.3 ± 9.81 881 ± 12.5 35800 ± 3460 35500 ± 3520 12.1 ± 2.05 - - 43.0 ± 4.17 p.o. 60 13 ± 9.2 947 ± 140 45300 ± 8860 45100 ± 8840 16 ± 0.61 - - 58a AUC0-24.10 ▐2014 Worldwide NCEsTable 6 In Vivo Pharmacokinetic Parameters of Ceritinib in Patients Following a Single Doses of Ceritinib on[5]50n = 1 n = 1 n = 1 n = 1 n = 1 n = 1 n = 1100 19.4n = 115.0(6.00-24.0)n = 229.3(10.1)n = 2467(10.7)n = 2938(24.3)n = 23250n = 1116n = 1200 33.2n = 15.08(4.17-6.00)n = 240.2(88.5)n = 2703(55.6)n = 21460(62.9)n = 23720n = 177.5n = 130030.1(10.0)n = 34.00(4.00-5.95)n = 3198(41.5)n = 33440(44.7)n = 37470(46.5)n = 31880(50.7)n = 244.5(36.8)n = 240030.7(39.1)n = 104.99(2.97-6.73)n = 12120(80.9)n = 121920(78)n = 124070(81.8)n = 123470(74.4)n = 595.9(58.6)n = 550031.1(11.1)n = 73.98(3.00-23.5)n = 8153(86.5)n = 82350(87.9)n = 85140(142)n = 86230(219)n = 3147(170)n = 360037.6(24.6)n = 66.00(3.00-24.1)n = 9212(59.7)n = 93590(53.4)n = 98180(57.4)n = 91990(4.30)n = 246.39.60)n = 270038.9(98.4)n = 36.00(4.00-25.0)n = 4206(146)n = 43450(138)n = 49210(112)n = 42340(56.5)n = 266.6(35.8)n = 275040.6(34.7)n = 96.02(3.95-23.8)n = 10186(127)n = 103390(121)n = 107870(127)n = 104230(164)n = 388.5(163)n = 3n: Number of patients with non-missing values. Values were median (range) for T max, geometric mean (CV% of geometric mean) for all others.Key finding:Ceritinib was classified as a low passive permeability compound.DistributionTable 7 In Vitro Plasma Protein Binding and Blood Partitioning of [14C]Ceritinib[5]Rat 97.9 ± 2 98.4 ± 0 98.4 ± 0.1 100 97.6 ± 0.6 Rat0.4 3.7 1.3Dog 98.8 ± 0.5 98.3 ± 2.1 98.4 ± 0.810000 98.6 ± 0.8 Dog66.6 ±1.667.0 ±0.471.6 ±1.1Monkey 94.4 ± 0.5 97.4 ± 0.4 95.2 ± 0.2 Monkey 78.7 ±1.177.8 ±1.576.4 ±0.4Human 98.8 ± 0.5 97.2 ± 1.4 96.7 ± 1.8 Human 58.2 ±3.558.6 ±5.556.9 ±4.8[4] FDA Datadase. /drugsatfda_docs/nda/2014/205755Orig1s000PharmR.pdf (accessed Nov 2015).14[4]Blood 63.4 600 220 48.0 NM NM 1.00 1420 1 Brain 29.8 42.0 20.0 13.0 NM NM 0.148 137 0.096 Bile NM 74000 29000 11000 NM NM 155 48800 34.4 Lungs 607 18000 5900 2100 28.9 21.2 37.2 23300 16.4 Liver 2720 33000 8100 1800 78.7 130 52.6 29500 20.8 Kidney cortex 693 17000 6400 1300 20.3 28.3 32.8 23700 16.7 Stomach glandular 13400 5200 1900 310 NM NM 26.3 6590 4.64 Adrenal cortex 1870 37000 11000 4100 41.4 NM 72.4 48700 34.4 Harderian gland 31.5 1700 1800 2600 1060 2990 62.8 5040 3.55 Bone marrow 198 **** **** 2700 23.3 NM 28.0 9220 6.50 Small intestine wall 164000 490000 11000 730 NM NM 413 6980 4.92 Thyroid gland 843 18000 7300 1200 NM NM 30.3 35100 24.7 Pituitary gland 229 14000 6600 15000 1810 2590 211 15000 10.6 Spleen 1040 27000 6400 2500 42.3 41.6 47.6 NA NA Lymph node 200 19000 2900 4100 183.0 NM 45.1 8720 6.14 Adrenal medulla 890 21000 2100 1300 48.1 NM 26.5 14600 10.3 Pancreas 388 13000 4000 1300 6.6 NM 24.6 14200 9.98 Thymus 120 4300 1400 2400 86.7 NM 21.3 3860 2.72 Salivary gland 208 9700 3500 1700 NM NM 20.2 8740 6.16 Uveal tract 108 6500 1100 8500 2880 8160 176 8600 6.06 NM: Not measured,Metabolism14[5]Rat 2.5 0-18 NC NC NCNC 12.5 0-24 NC NC NCMonkey 2.5 0-4 7.02 98.7 312.90.12 12.5 0-8 18.0 38.5 131.7Human 2.5 0.25-18 100.4 6.90 17.30.014 12.5 0-18 66.95 10.4 25.8NC: Not calculated.[4] FDA Datadase. /drugsatfda_docs/nda/2014/205755Orig1s000PharmR.pdf (accessed Nov 2015).14[6]Ketoconazole (3A) 0-1 0.100 ~0.03 90.5 Azamulin (3A) 0-5 0.150 ~0.01 100 Ticlopidine (2B6/2C19) 0-10 1.70 >10 10.9S-mephenytoin (2C19) 0-250 95.5 >250 18 Montelukast (2C8) 0-2 0.0140 (0.18-0.71) >2 16.5 Furafylline (1A2) 0-10 2.00 >10 19.1 Quinidine (2D6) 0-1 0.0605 >1 17.9 Sulfaphenazole (2C9) 0-5 0.510 >5 11.9 Fluvastatin (2C9) 0-5 0.525 >5 21.6a Median value were calculated from (DMPK R0400785).If a range of values was reported for a study an average value was taken. The K i or IC50 values used in the calculations corresponded to inhibition of the enzyme (or subfamily, in the case of CYP3A).b Percent maximal inhibition of total [14C]Ceritinib metabolism at the concentrations of inhibitor examined.Rat a Ceritinib 108.03 ± 2.34 324.73 ± 17.09 426.58 ± 62.86 609.30 ± 32.39 L746530 168.29 ± 1.3 460.89 ± 22.3 709.78 ± 43.0 1068.22 ± 17.5Human a Ceritinib 105.80 ± 7.02 281.74 ± 0.68 504.46 ± 16.86 702.25 ± 57.12 L746530 173.98 ± 8.8 523.11 ± 0.5 684.59 ± 70.4 1057.12 ± 76.3Rat b Ceritinib 11.59 ± 0.25 34.73 ± 1.83 45.62 ± 6.72 65.17 ± 3.46 L746530 18.00 ± 0.14 49.29 ± 2.38 75.91 ± 4.60 114.25 ± 1.88Human b Ceritinib 11.32 ± 0.75 30.13 ± 0.07 53.95 ± 1.80 75.11 ± 6.11 L746530 18.61 ± 0.94 55.95 ± 0.05 73.22 ± 7.53 113.06 ± 8.16a Based on viable cells.b Based on 107 × 106 hepatocytes per gram of tissue (Wilson, et at 2003). Values were mean ± rang (n = 2) and rounded to three significant figures or to the nearest 0.01.[6]CeritinibRat 10.0 ± 1.2 21.2 ± 1.3 21.4 ± 0.9 15.0 ± 1.9 Human 10.4 ± 1.3 20.5 ± 0.5 12.3 ± 10.7 11.3 ± 0.9L746530Rat 64.4 ± 1.7 715.9 ± 35.7 579.8 ± 35.0 590.0 ± 49.5 Human 52.8 ± 2.6 254.3 ± 5.2 245.0 ± 24.1 354.1 ± 33.8a Based on nominal microsomal protein included in incubation. Values were mean ± SD (n = 3). All values were rounded to three significant figures or to the nearest 0.1.Monkey 2.50.25 92.5 0.23 0.31 ND 3.33 ND 0.68 1.20 ND ND0.5 89.9 0.28 0.59 ND 4.68 ND 0.77 1.77 ND ND1 83.8 0.81 0.95 ND 8.67 ND 0.58 3.58 ND ND2 76.9 1.96 1.26 ND 10.6 1.00 5.15 ND ND ND4 69.1 2.37 2.13 2.20 13.4 1.46 7.15 ND ND ND8 61.5 2.97 3.01 4.27 14.8 2.69 8.12 ND ND ND18 50.6 4.06 2.40 5.11 20.4 2.68 8.21 ND 2.51 2.9124 32.0 7.11 4.42 11.5 25.0 5.23 8.77 ND 2.15 2.5 12.50.25 NA NA NA NA NA NA NA NA NA NA0.5 92.7 0.23 0.24 ND 1.95 0.20 0.37 1.92 ND ND1 89.3 0.34 0.56 ND 3.11 0.41 0.53 3.09 ND ND2 80.7 1.68 0.97 1.09 6.05 1.17 5.60 ND ND ND4 80.6 1.12 1.21 1.56 5.32 1.81 6.12 ND ND ND8 74.2 2.16 1.34 4.06 5.96 3.48 5.90 ND ND 0.9318 71.2 2.10 1.43 5.61 6.81 3.34 5.07 ND 0.89 1.6924 61.9 4.05 1.81 8.11 9.31 5.82 5.41 ND 0.87 1.40Human 2.5 0.25 90.4 - - - 0.78 - 0.75 1.76 - ND0.5 90.9 - - - 0.77 - 0.98 1.19 - ND1 91.8 - - - 1.28 - 1.35 1.14 - ND2 91.6 - - - 2.22 - 1.18 1.02 - ND 4 89.9 - - - 3.11 - 1.46 1.26 - ND 8 87.6 - - - 4.52 - 1.47 1.55 - 1.11 18 83.2 - - - 8.17 - 1.61 1.52 - 1.59 24 81.4 - - - 8.94 - 2.62 1.38 - 2.28Human 12.5 0.25 NA - - - ND - NA NA - NA0.5 96.4 - - - ND - ND 3.60 - ND1 95.5 - - - ND - ND 4.47 - ND2 95.5 - - - ND - ND 4.55 - ND 4 95.7 - - - ND - ND 4.26 - ND 8 95.2 - - - ND - ND 4.81 - ND 18 85.1 - - - 7.05 - ND 7.89 - ND 24 90.8 - - - 2.96 - 3.93 0.67 - 1.65-: Not observed.Table 14 In Vivo Metabolites Profiles in Plasma, Bile and Feces of Rats, Monkeys and Humans[6]Plasma aRat i.v./p.o. 10/25 100 - - - - - - - - - - Monkeyi.v. 10 89.9 1.4 - 1.8 0.6 1.2 - 3 ---p.o. 30 84.4 3.6 - 2.5 0.4 1.6 - 3.1 ---Human p.o. 750 mg 81.6 - 1.2 1.4 1.9 1.6 1.8 2.3 1.7 1.7 2Bile Rat i.v. 10 34.89 ND ND - 1.09 3.26 0.83 ----p.o. 25 9.19 1.71 0.84 - 0.72 1.46 0.52 ----FecesRati.v. 10 12.05 3.75 4.56 - ND ND ND ----p.o. 25 51.77 4.54 5.88 - ND 2.82 ND ----Monkeyi.v. 10 55.1 1.8 2.8 - 3.7 10.9 17.9 0.3 ---p.o. 30 60.2 2.7 2.2 - 2.7 5.9 8.7 0.4 ---Human p.o. 750 mg 68 - 3.9 - 1.8 1.4 6.5 1.6 1.1 1.4 2.2M0: Ceritinib. a %AUC for metabolites in plasma.Figure D Proposed Pathways for In Vivo Biotransformation of [14C]Ceritinib in Rats, Monkeys and Humans[6] The red labels represent the major components in matrices. P = Plasma, U = Urine, B = Bile, F = Feces.ExcretionRat (intact)10 i.v. 0-168 - 0.241 ± 0.174 107 ± 7.825 p.o. 0-168 - 0.180 101 Rat (BDC)10 i.v. 0-72/168 65.4 0.62 29.825 p.o. 0-72/168 24.3 ± 8.52 1.05 ± 1.03 65.0 ± 17.0 Monkey10 i.v. 0-168 - 0.59 10530 p.o. 0-168 - 0.71 ± 0.45 92.3 ± 6.94 Human 9.14 p.o. 0-168 - 1.2 91.0Drug-Drug Interaction[5, 6]CYP1A2 Phenacetin >100 (2.5) - 1.0CYP1A2 mRNA 1.01 1.14 1.07 32.1CYP2A6 Coumarin 5 (1.5) 0.03 (0.009) 61.0 Activity 1.15d 1.22 1.01 31.7CYP2B6 Bupropion 2 (0.3) 5.3 (0.780) 1.3CYP2B6 mRNA 1.02 1.17 0.96 10.2CYP2C8 Paclitaxel 25 (0.6) - 1.1 Activity 1.05 1.20 0.92 4.65CYP2C8 Amodiaquine 2 (0.6) 16.7 (4.86) 1.1CYP2C9 mRNA 1.12 1.15 1.18 2.34CYP2C9 Diclofenac 2 (0.6) 0.24 (0.0701) 8.5 Activity 1.27 1.77 1.10 3.50CYP2C19 S-mephenytoin 70 (1.8) - 1.0CYP3A4 mRNA 1.27 2.73 6.03b31.1CYP2D6 Bufuralol 20 (2.9) - 1.2 Activity 0.98 1.43 1.33c8.81 CYP2E1 Chlorzoxazone 30 (4.4) - 1.1CYP3A4/5 Midazolam 0.2 (0.06) 0.16 (0.0469) 12.3CYP3A4/5 Testosterone 0.2 (0.06) -19.0Calculation of R1 values based on maximal steady-state concentration of 1010 ng/mL or 1.8 μM. [I]/K i assumed to be IC50/2 for competitive inhibition for those CYP enzymes without an experimental K i value. a Omeprazole 50 μM for CYP1A2, phenobarbital 1000 μM for CYP2B6 and CYP2C9, rifampin 10 μM for CYP3A4. b Mean of 3.24-, 8.74-, and 6.12-fold change relative to vehicle control in the three human hepatocyte lots. c Mean of 0.936-, 0.983-, 2.06-fold change relative to vehicle control in the three human hepatocyte lots. d:Mean of 1.75-, 1.24-, and 2.31-fold change relative vehicle control in the three human hepatocyte lots.[5]。
看不懂基因检测报告?实用科普一文带你全搞定!

看不懂基因检测报告?实⽤科普⼀⽂带你全搞定!⾃从肺癌遇到靶向药物,就给肺癌的治疗打开⼀扇新的⼤门。
要选择合适的或者正确的靶向药物,⾮常重要的⼀点就是做基因检测,基因检测是指导靶向药的选择和进⾏个性化治疗必不可缺的步骤。
但是,很多觅友都会有这样⼀个问题,拿着基因检测报告单却看不懂!上⾯密密⿇⿇的英⽂字母都是什么意思?我应该选择哪⼀种靶向药?科普君综合了⼏份基因检测报告来给⼤家解读⼀下,如何从基因检测报告中看到有效信息。
肿瘤基因检测报告从结构上⼤部分可以分为三部分:1、患者基本信息及标本信息;2、患者的基因检测结果;3、靶点筛药:适⽤的药物和局限性由于每个医院或基因检测机构的报告单样式不⼀样,会有⼀些的项⽬增加或删减,但我们要从这⾥获取主要的信息是:●我的基因检测结果是阴性还是阳性?●是哪⼀个基因产⽣了突变?●这个基因⽬前有可⽤的靶向药物吗?基因检测结果是阴性还是阳性?N C C N⾮⼩细胞肺癌临床实践指南(2018.V1)中对于“分⼦诊断与靶向治疗原则”(N S C L-G)明确指出基因检测时⼀定要包含这8种基因靶点:E G F R、A LK、R E T、R O S1、M E T、E R B B2、K R A S、B R A F根据检测结果或变异结果,就可以很直观的看出是否发⽣了基因突变。
变异结果如果出现“+”、或显⽰具体基因则说明存在该种突变类型;如果是出现“野⽣型”、"-"、“未检出”则说明未发现该基因突变。
从上⾯的表格中,我们就可以看到基因检测的结果的是“未检测到”,且⼋个基因靶点都是“未检测到”,就说明患者的基因突变是阴性的。
有的基因检测报告会有⽂字直接说明,“本次检测未检出相关的基因变异”这类字样。
阳性的基因突变会在检测结果中直接显⽰是哪⼀种基因产⽣了突变,下图就显⽰的是E GF R突变,且突变频率为0.44%。
基因的变异类型是什么意思?我的突变类型有药可⽤吗?1.厄洛替尼(特罗凯)(E r lot inib)--第⼀代药物⼝服150m g,每⽇⼀次。
奥希替尼分子式

奥希替尼分子式
奥希替尼(Axitinib)是一种治疗肾细胞癌的靶向药物。
它的分子式为C22H18N4OS,是一种小分子酪氨酸激酶抑制剂。
奥希替尼通过抑制肿瘤血管生成和生长来发挥治疗作用。
奥希替尼是一种口服药物,它能够选择性地靶向肿瘤细胞中的血管内皮生长因子受体(VEGFR)家族。
这些受体在肿瘤的血管生成过程中起着重要的作用。
奥希替尼通过与VEGFR结合,阻断了肿瘤细胞的血管生成信号通路,从而抑制了肿瘤的生长和扩散。
奥希替尼的治疗效果已经在临床试验中得到证实。
研究表明,奥希替尼可以显著延长肾细胞癌患者的生存期,并减少肿瘤的进展风险。
此外,奥希替尼还可以缓解患者的疼痛和改善生活质量。
尽管奥希替尼在治疗肾细胞癌方面取得了很大的成功,但它也存在一些不良反应。
常见的不良反应包括高血压、蛋白尿、口腔溃疡、手足综合征等。
这些不良反应大多可以通过调整剂量或辅助治疗来控制。
总的来说,奥希替尼是一种有效的治疗肾细胞癌的药物。
它通过靶向肿瘤血管生成信号通路,抑制肿瘤的生长和扩散。
尽管存在一些不良反应,但奥希替尼的治疗效果已经得到证实。
对于肾细胞癌患者来说,奥希替尼提供了一种新的治疗选择,可以延长生存期并改善生活质量。
2021最新抗肿瘤药物分级目录31

V
谈判药品医保限定
安罗替尼
Anlotinib
胶囊
V
谈判药品 医保限定
索拉非尼Sorafenib
片剂
V
谈判药品医保限定
瑞戈非尼
Regorafenib
片剂
V
谈判药品 医保限定
仑伐替尼
Lenvatinib
胶囊
V
谈判药品医保限定
阿帕替尼
Apatinib
片剂
V
谈判药品 医保限定
舒尼替尼Sunitinib
胶囊
r:保限定
尼洛替尼
Nilotinib
g g
胶囊
V
达沙替尼Dasatinib
片剂
V
医保限定
氟马替尼
Flumatinib
片剂
V
诔判药品 医保限定
伊布替尼Ibrutinib
胶囊
V
彳豹鞋岛
泽布替尼Zanubrutinib
胶囊
V
谈判药品!'.'1<i
二、分子靶
芦可替尼Ruxolitinib
片剂
V
向治疗药
物
吉非替尼Gefitinib
伊尼妥单抗Inetetamab
针剂
V
谈判药品
拉帕替尼Lapatinib
片剂
V
非医保
毗咯替尼Pyrotinib
片剂
V
谈判药品
奈拉提尼Neratinib
片剂
V
非医保
哌柏西利Palbociclib
胶囊
V
非医保
西妥昔单抗Cetuximab
汴射液
V
谈判药品 医保限定
尼妥珠单抗
CSCO大家访谈录︱杨衿记教授:ASCEND-8临床研究的启示和意义

CSCO⼤家访谈录︱杨衿记教授:ASCEND-8临床研究的启⽰和意义杨衿记教授编者按:晚期⾮⼩细胞肺癌(NSCLC)的靶向药物治疗,是精准医疗的最佳典范。
靶向药物的最佳治疗剂量是在疗效和毒性之间寻找到的最佳平衡点,ASCEND-8临床研究基于前期研究中⼆代ALK抑制剂塞瑞替尼所出现的毒性反应,创新性地探索、对⽐了调整剂量和⽤药⽅式后的疗效和毒性,研究发现:塞瑞替尼450mg随餐服⽤的⽅式较以往750mg空腹⼝服的⽅式不良反应更低,⽽疗效也有增加的趋势,⽬前,塞瑞替尼450mg随餐给药⽅式⼀线中位⽆进展⽣存(PFS)经第三⽅评估已经超过25个⽉,仍未达到。
这⼀研究给临床医⽣和新药研发带来了哪些启⽰和意义呢?本届CSCO⼤会中,《肿瘤瞭望》⼩编⾮常荣幸地邀请到肺癌届专家⼴东省⼈民医院杨衿记教授来为⼤家解答这⼀问题。
2019 CSCO《肿瘤瞭望》:在早期ASENCD-4临床研究中,塞瑞替尼⼀线治疗采⽤的是750mg空腹给药的⽤药⽅式,⽽⽬前中国塞瑞替尼的获批⽅案是450mg随餐服⽤,这⼀剂量调整是基于ASCEND-8研究结果,您在临床⼯作中对于塞瑞替尼调整剂量的应⽤有何体会?2019 CSCO杨衿记教授:这个问题⾮常好。
塞瑞替尼作为⼆代ALK抑制剂,研发路程实现了精准化,ASCEND-4临床研究中,塞瑞替尼750mg空腹服⽤⼀线治疗ALK阳性晚期NSCLC的疗效、 PFS均明显优于化疗,但是呕吐等胃肠道不良事件的发⽣率较⾼,甚⾄超过了⼀代药物克唑替尼,副作⽤⽅⾯是临床医⽣⼼中挥之不去的阴影。
但是,ASCEND-8研究将塞瑞替尼调整为450mg随餐服⽤的⽅式后,毒性反应⼤⼤降低,75%以上的患者基本不会出现3/4级严重不良反应,个别不良反应发⽣率也和其他TKI⽆异。
因此,塞瑞替尼450mg随餐⼝服⽆论是作为⼀线还是⼆线治疗,都是⾮常好的选择,不仅疗效较750mg剂量未降低,⼜能够提⾼患者⽣活质量、减轻毒性,令患者得到获益的同时,也⽅便临床医⽣、护理⼈员观察毒性反应。
ritlecitinib结构

ritlecitinib结构
【实用版】
目录
1.瑞替尼布的结构概述
2.瑞替尼布的结构特征
3.瑞替尼布的作用机制
4.瑞替尼布的应用领域
5.瑞替尼布的研究进展
正文
1.瑞替尼布的结构概述
瑞替尼布(Ritlecitinib)是一种新型的靶向抗肿瘤药物,属于抑制剂类,具有很高的治疗效果。
它的化学名称为 N-[4-[[3-(2-甲氧基乙氧基)-5-(三氟甲基)-2-吡啶] 胺基]-7-氧代 -6-氮杂 -2-脱氧-α-L-核糖 -9-芴甲酸],是一种小分子化合物,具有很好的水溶性。
2.瑞替尼布的结构特征
瑞替尼布的结构特征主要包括以下几个方面:首先,它含有一个吡啶环,这个环上有一个三氟甲基取代基;其次,它还含有一个氧代芴环,这个环上有一个甲氧基取代基;最后,它的侧链上还有一个氧原子,这个原子与核糖环上的一个碳原子形成一个醚键。
3.瑞替尼布的作用机制
瑞替尼布通过抑制肿瘤细胞的生长和增殖来发挥其抗肿瘤作用。
它主要作用于肿瘤细胞的某些关键信号通路,如 PI3K/AKT 信号通路、
RAS/RAF/MEK 信号通路等,从而阻断这些信号通路的正常功能,导致肿瘤细胞死亡。
4.瑞替尼布的应用领域
瑞替尼布主要用于治疗各种实体瘤,如肺癌、乳腺癌、结直肠癌等。
此外,它还可以用于治疗血液肿瘤,如急性髓系白血病、慢性髓系白血病等。
5.瑞替尼布的研究进展
近年来,瑞替尼布在抗肿瘤领域得到了广泛的关注。
许多研究表明,瑞替尼布具有很好的抗肿瘤活性,并且在临床试验中取得了显著的疗效。
赛可瑞 (Ceritinib)使用说明书

赛可瑞 (Ceritinib)使用说明书赛可瑞 (Ceritinib)使用说明书一、药物简介赛可瑞 (Ceritinib)是一种口服抗癌药物,属于酪氨酸激酶抑制剂。
其主要成分是赛可瑞 (Ceritinib)。
本药物适用于特定类型的非小细胞肺癌 (NSCLC) 患者,具有抑制肿瘤生长和扩散的作用。
二、适应症赛可瑞 (Ceritinib)适用于以下情况的患者:1. 未曾接受过其他治疗方法,具体为 ROS1 长序列擬胞嘧啶酸檢出陽性的局部晚期或已转移性非小细胞肺癌患者。
2. 针对曾接受过铂类化疗后疾病进展的ALK 阳性转移性非小细胞肺癌患者。
三、使用方法1. 用药时间:每天服用1次。
2. 用药剂量:成人每次口服450毫克。
3. 是否与饭前或饭后一起服用:药物可以与或者不配合进餐一起服用,根据患者个体差异,不同患者可自行选择。
然而,应当避免与高脂餐同时服用,因为高脂餐可导致赛可瑞的血浆药物浓度升高。
4. 忘记用药:如果忘记一次剂量,可以在下一次计划剂量服用时正常使用,不需要补充遗漏的剂量。
四、禁忌以下情况下禁止使用赛可瑞 (Ceritinib):1. 对赛可瑞 (Ceritinib)成分过敏的患者。
2. 使用赛可瑞 (Ceritinib)期间同时进行心脏电图监测时,出现心电图 QT 间期延长,或异常心脏情况,如心脏骤停、心功能不全等。
五、注意事项1. 肝脏功能:使用赛可瑞 (Ceritinib)时,应特别注意对肝脏功能的监测。
如果出现肝功能异常或肝损害症状(如黄疸、乏力等),应立即停用赛可瑞 (Ceritinib)。
2. 心脏功能:在使用赛可瑞 (Ceritinib)期间出现心脏功能障碍、不齐和 QT 间期延长等异常情况,需减少或停用赛可瑞 (Ceritinib)。
应对患者进行心脏电图监测,以确保患者的心脏健康状况。
3. 妊娠和哺乳期:赛可瑞 (Ceritinib)可能对胎儿造成危害,因此对于妊娠期或哺乳期妇女,应权衡利弊,并在医生指导下使用。
ALK阳性晚期肺癌的靶向治疗
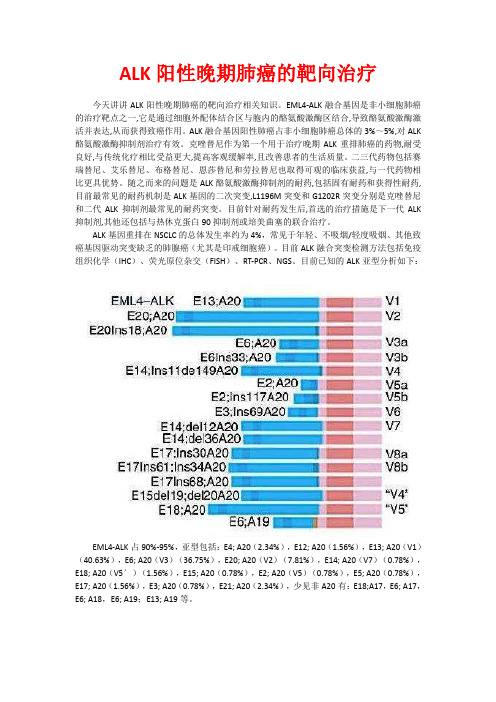
ALK阳性晚期肺癌的靶向治疗今天讲讲ALK阳性晚期肺癌的靶向治疗相关知识。
EML4-ALK融合基因是非小细胞肺癌的治疗靶点之一,它是通过细胞外配体结合区与胞内的酪氨酸激酶区结合,导致酪氨酸激酶激活并表达,从而获得致癌作用。
ALK融合基因阳性肺癌占非小细胞肺癌总体的3%~5%,对ALK 酪氨酸激酶抑制剂治疗有效。
克唑替尼作为第一个用于治疗晚期ALK重排肺癌的药物,耐受良好,与传统化疗相比受益更大,提高客观缓解率,且改善患者的生活质量。
二三代药物包括赛瑞替尼、艾乐替尼、布格替尼、恩莎替尼和劳拉替尼也取得可观的临床获益,与一代药物相比更具优势。
随之而来的问题是ALK酪氨酸激酶抑制剂的耐药,包括固有耐药和获得性耐药,目前最常见的耐药机制是ALK基因的二次突变,L1196M突变和G1202R突变分别是克唑替尼和二代ALK抑制剂最常见的耐药突变。
目前针对耐药发生后,首选的治疗措施是下一代ALK 抑制剂,其他还包括与热休克蛋白90抑制剂或培美曲塞的联合治疗。
ALK基因重排在NSCLC的总体发生率约为4%,常见于年轻、不吸烟/轻度吸烟、其他致癌基因驱动突变缺乏的肺腺癌(尤其是印戒细胞癌)。
目前ALK融合突变检测方法包括免疫组织化学(IHC)、荧光原位杂交(FISH)、RT-PCR、NGS。
目前已知的ALK亚型分析如下:EML4-ALK占90%-95%,亚型包括:E4; A20(2.34%),E12; A20(1.56%),E13; A20(V1)(40.63%),E6; A20(V3)(36.75%),E20; A20(V2)(7.81%),E14; A20(V7)(0.78%),E18; A20(V5′)(1.56%),E15; A20(0.78%),E2; A20(V5)(0.78%),E5; A20(0.78%),E17; A20(1.56%),E3; A20(0.78%),E21; A20(2.34%),少见非A20有:E18;A17,E6; A17,E6; A18,E6; A19;E13; A19等。
- 1、下载文档前请自行甄别文档内容的完整性,平台不提供额外的编辑、内容补充、找答案等附加服务。
- 2、"仅部分预览"的文档,不可在线预览部分如存在完整性等问题,可反馈申请退款(可完整预览的文档不适用该条件!)。
- 3、如文档侵犯您的权益,请联系客服反馈,我们会尽快为您处理(人工客服工作时间:9:00-18:30)。
呼吸系统肿瘤用药塞瑞替尼 ceritinib
制剂与规格:胶囊:150mg
适应证:本品适用于此前接受过克唑替尼治疗后进展的或者对克唑替尼不耐受的ALK阳性的局部晚期或转移性NSCLC患者。
合理用药要点:
1.用药前必须明确有经国家药品监督管理局批准的间变性淋巴瘤激酶检测方法检测到的ALK阳性。
2.本品的推荐剂量为每日一次,每次450mg,每天在同一时间口服给药,药物应与食物同时服用。
根据患者个体的安全性或耐受性情况,在治疗过程中可能需要暂时中断使用本品或下调剂量,应以150mg的下调幅度逐渐减少本品的日剂量。
应注意早期识别药物不良反应并及早给予标准的支持性治疗措施。
对于无法耐受每日随餐服用150mg剂量的患者,应停用本品。
3.用药期间须注意胃肠道不良反应、肝毒性、间质性肺炎/非感染性肺炎、心律失常、高血糖等不良反应。
4.本品治疗期间应避免联合使用强效CYP3A抑制剂。
如果必须同时使用强效CYP3A抑制剂,则应将塞瑞替尼的剂量减少约三分之一,取整至最接近的150mg整数倍剂量。
5.如果本品与P-gp抑制剂联合使用,可能导致本品浓度
1。